Фильтры Блума в биоинформатике
Фильтры Блума с эффективным использованием пространства, — это вероятностные структуры данных используемые для проверки того, является ли элемент частью набора. Фильтры Блума требуют гораздо меньше места, чем другие структуры данных для представления наборов, однако недостатком фильтров Блума является вероятность ложных срабатываний при запросе структуры данных. Поскольку несколько элементов могут иметь одинаковые хэш-значения для нескольких хэш-функций, существует вероятность того, что запрос несуществующего элемента может вернуть положительный результат, если в фильтр Блума был добавлен другой элемент с такими же хеш-значениями. Предполагая, что хеш-функция имеет равную вероятность выбора любого индекса фильтра Блума, частота ложных срабатываний при запросе фильтра Блума является функцией количества бит, количества хеш-функций и количества элементов фильтра Блума. Это позволяет пользователю управлять риском получения ложноположительного результата за счет снижения пространственных преимуществ фильтра Блума.
Фильтры Блума в основном используются в биоинформатике для проверки существования k-мера в последовательности или наборе последовательностей. K-меры последовательности индексируются в фильтре Блума, и любой k-мер того же размера можно запросить с помощью фильтра Блума. Это предпочтительная альтернатива хешированию k-меров последовательности с помощью хеш-таблицы , особенно когда последовательность очень длинная, поскольку хранение большого количества k-меров в памяти требует больших затрат.
Приложения
[ редактировать ]Характеристика последовательности
[ редактировать ]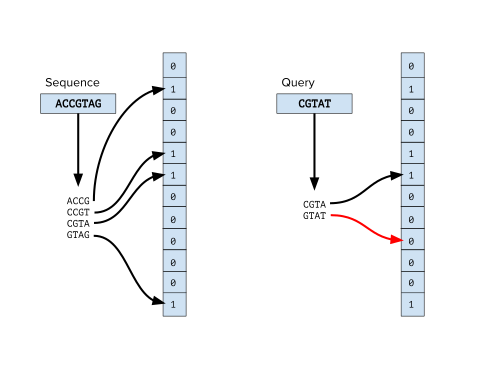
Шаг предварительной обработки во многих приложениях биоинформатики включает классификацию последовательностей, в первую очередь классификацию считываний из эксперимента по секвенированию ДНК . Например, в метагеномных исследованиях важно иметь возможность определить, принадлежит ли результат секвенирования новому виду. [1] а в проектах клинического секвенирования жизненно важно отфильтровывать считывания геномов загрязняющих организмов. Существует множество инструментов биоинформатики, которые используют фильтры Блума для классификации прочтений путем запроса k-меров прочтения к набору фильтров Блума, сгенерированных на основе известных эталонных геномов . Некоторые инструменты, использующие этот метод, — FACS. [2] и инструменты BioBloom. [3] Хотя эти методы не могут превзойти другие инструменты классификации биоинформатики, такие как Kraken, [4] они предлагают альтернативу с эффективным использованием памяти.
Недавняя область исследований с использованием фильтров Блума для характеристики последовательностей заключается в разработке способов запроса необработанных показаний экспериментов по секвенированию. Например, как можно определить, какие чтения содержат конкретный 30-мер во всем архиве чтения последовательностей NCBI ? Эта задача аналогична той, которую выполняет BLAST , однако она включает в себя запрос к гораздо большему набору данных; в то время как BLAST запрашивает базу данных эталонных геномов, эта задача требует, чтобы возвращались конкретные чтения, содержащие k-мер. BLAST и подобные инструменты не могут эффективно справиться с этой проблемой, поэтому для этой цели были реализованы структуры данных на основе фильтра Блума. Бинарные цветущие деревья [5] представляют собой бинарные деревья фильтров Блума, которые облегчают запрос транскриптов в крупных экспериментах по секвенированию РНК . БИГСИ [6] заимствует побитовые подписи из области поиска документов для индексации и запроса всех данных микробного и вирусного секвенирования в Европейском архиве нуклеотидов . Подпись данного набора данных кодируется как набор фильтров Блума из этого набора данных.
Сборка генома
[ редактировать ]
Эффективность памяти фильтров Блума использовалась при сборке генома как способ уменьшить объем занимаемого k-мерами данных секвенирования. Вклад методов сборки на основе фильтров Блума заключается в объединении фильтров Блума и графов де Брейна в структуру, называемую вероятностным графом де Брейна. [7] который оптимизирует использование памяти за счет частоты ложных срабатываний, свойственной фильтрам Блума. Вместо хранения графа де Брейна в хеш-таблице он сохраняется в фильтре Блума.
Использование фильтра Блума для хранения графа де Брёйна усложняет этап обхода графа для построения сборки, поскольку информация о ребрах не кодируется в фильтре Блума. Обход графа осуществляется путем запроса к фильтру Блума любого из четырех возможных последующих k-меров из текущего узла. Например, если текущий узел предназначен для k-мера ACT, то следующий узел должен быть для одного из k-меров CTA, CTG, CTC или CTT. Если в фильтре Блума существует k-мер запроса, то к пути добавляется k-мер. Таким образом, существует два источника ложных срабатываний при запросе фильтра Блума при обходе графа де Брейна. Существует вероятность того, что один или несколько из трех ложных k-меров существуют где-то в наборе секвенирования, чтобы дать ложноположительный результат, а также существует вышеупомянутая частота ложноположительных результатов, присущая самому фильтру Блума. Инструменты сборки, использующие фильтры Блума, должны учитывать эти источники ложных срабатываний в своих методах. АБИСС 2 [8] и Миниа [9] являются примерами ассемблеров, использующих этот подход для сборки de novo .
Исправление ошибок секвенирования
[ редактировать ]Методы секвенирования нового поколения (NGS) позволили генерировать новые последовательности генома намного быстрее и дешевле, чем предыдущие методы секвенирования Сэнгера . Однако эти методы имеют более высокий процент ошибок, [10] [11] что усложняет последующий анализ последовательности и даже может привести к ошибочным выводам. Было разработано множество методов для исправления ошибок чтения NGS, но они используют большие объемы памяти, что делает их непрактичными для больших геномов, таких как геном человека. Поэтому для устранения этих ограничений были разработаны инструменты, использующие фильтры Блума, за счет эффективного использования памяти. Мушкет [12] и БЛАГОСЛОВИТЕ [13] являются примерами таких инструментов. Оба метода используют подход спектра k-меров для исправления ошибок. Первым шагом этого подхода является подсчет множественности k-меров, однако в то время как BLESS использует только фильтры Блума для хранения счетчиков, Musket использует фильтры Блума только для подсчета уникальных k-меров и сохраняет неуникальные k-меры в хеш-таблица, как описано в предыдущей работе [14]
РНК-Seq
[ редактировать ]Фильтры Блума также используются в некоторых конвейерах RNA-Seq . РНК-ским [15] кластеризует транскрипты РНК, а затем использует фильтры Блума для поиска сиг-меров: k-меров, которые встречаются только в одном из кластеров. Эти сиг-меры затем используются для оценки уровней содержания транскриптов. Следовательно, он не анализирует все возможные k-меры, что приводит к улучшению производительности и использования памяти, и было показано, что он работает так же хорошо, как и предыдущие методы.
Ссылки
[ редактировать ]- ^ Лундеберг, Йоаким; Арвестад, Ларс; Андерссон, Бьёрн; Алландер, Тобиас; Келлер, Макс; Страннехайм, Хенрик (1 июля 2010 г.). «Классификация последовательностей ДНК с использованием фильтров Блума» . Биоинформатика . 26 (13): 1595–1600. doi : 10.1093/биоинформатика/btq230 . ISSN 1367-4803 . ПМК 2887045 . ПМИД 20472541 .
- ^ Лундеберг, Йоаким; Арвестад, Ларс; Андерссон, Бьёрн; Алландер, Тобиас; Келлер, Макс; Страннехайм, Хенрик (1 июля 2010 г.). «Классификация последовательностей ДНК с использованием фильтров Блума» . Биоинформатика . 26 (13): 1595–1600. doi : 10.1093/биоинформатика/btq230 . ISSN 1367-4803 . ПМК 2887045 . ПМИД 20472541 .
- ^ Чу, Джастин; Садеги, Сара; Раймонд, Энтони; Джекман, Шон Д.; Нип, Ка Мин; Мар, Ричард; Мохамади, Хамид; Баттерфилд, Ярон С.; Робертсон, А. Гордон (1 декабря 2014 г.). «Инструменты BioBloom: быстрый, точный и экономичный скрининг последовательностей видов-хозяев с использованием фильтров Блума» . Биоинформатика . 30 (23): 3402–3404. doi : 10.1093/биоинформатика/btu558 . ISSN 1367-4811 . ПМК 4816029 . ПМИД 25143290 .
- ^ Вуд, Деррик Э.; Зальцберг, Стивен Л. (3 марта 2014 г.). «Кракен: сверхбыстрая классификация метагеномных последовательностей с использованием точного выравнивания» . Геномная биология . 15 (3): Р46. дои : 10.1186/gb-2014-15-3-r46 . ISSN 1474-760X . ПМК 4053813 . ПМИД 24580807 .
- ^ Карл Кингсфорд; Соломон, Брэд (март 2016 г.). «Быстрый поиск тысяч коротких экспериментов по секвенированию» . Природная биотехнология . 34 (3): 300–302. дои : 10.1038/nbt.3442 . ISSN 1546-1696 . ПМЦ 4804353 . ПМИД 26854477 .
- ^ Икбал, Замин; Маквин, Гил; Роша, Эдуардо ПК; Баккер, Хенк К. ден; Брэдли, Фелим (февраль 2019 г.). «Сверхбыстрый поиск всех депонированных бактериальных и вирусных геномных данных» . Природная биотехнология . 37 (2): 152–159. дои : 10.1038/s41587-018-0010-1 . ISSN 1546-1696 . ПМК 6420049 . ПМИД 30718882 .
- ^ Браун, К. Титус; Тидже, Джеймс М.; Хау, Адина; Канино-Конинг, Розангела; Хинтце, Аренд; Пелл, Джейсон (14 августа 2012 г.). «Масштабирование сборки последовательности метагенома с помощью вероятностных графов де Брёйна» . Труды Национальной академии наук . 109 (33): 13272–13277. arXiv : 1112.4193 . Бибкод : 2012PNAS..10913272P . дои : 10.1073/pnas.1121464109 . ISSN 0027-8424 . ПМК 3421212 . ПМИД 22847406 .
- ^ Бироль, Инанк; Уоррен, Рене Л.; Кумб, Лорен; Хан, Хамза; Джахеш, Гольназ; Хаммонд, С. Остин; Да, Сара; Чу, Джастин; Мохамади, Хамид (01 мая 2017 г.). «ABySS 2.0: ресурсоэффективная сборка больших геномов с использованием фильтра Блума» . Геномные исследования . 27 (5): 768–777. дои : 10.1101/гр.214346.116 . ISSN 1088-9051 . ПМК 5411771 . ПМИД 28232478 .
- ^ Чихи, Райан; Ризк, Гийом (16 сентября 2013 г.). «Экономичное и точное представление графа де Брейна на основе фильтра Блума» . Алгоритмы молекулярной биологии . 8 (1): 22. дои : 10.1186/1748-7188-8-22 . ISSN 1748-7188 . ПМЦ 3848682 . ПМИД 24040893 .
- ^ Ломан, Николас Дж.; Мисра, Раджу В.; Даллман, Тимоти Дж.; Константиниду, Кристала; Гарбия, Сахир Э.; Уэйн, Джон; Паллен, Марк Дж. (май 2012 г.). «Сравнение производительности настольных платформ высокопроизводительного секвенирования». Природная биотехнология . 30 (5): 434–439. дои : 10.1038/nbt.2198 . ISSN 1546-1696 . ПМИД 22522955 . S2CID 5300923 .
- ^ Ван, Синь Виктория; Блейдс, Натали; Дин, Цзе; Султана, Разван; Пармиджани, Джованни (30 июля 2012 г.). «Оценка частоты ошибок секвенирования при коротких чтениях» . БМК Биоинформатика . 13 :185. дои : 10.1186/1471-2105-13-185 . ISSN 1471-2105 . ПМЦ 3495688 . ПМИД 22846331 .
- ^ Шмидт, Бертиль; Шредер, Ян; Лю, Юнчао (01 февраля 2013 г.). «Мушкет: многоступенчатый корректор ошибок на основе спектра k-меров для данных последовательности Illumina» . Биоинформатика . 29 (3): 308–315. doi : 10.1093/биоинформатика/bts690 . ISSN 1367-4803 . ПМИД 23202746 .
- ^ Ху, Вэнь-Мэй; Ма, Цзянь; Чен, Деминг; Ву, Сяо-Лун; Хо, Юн (15 мая 2014 г.). «BLESS: решение для коррекции ошибок на основе фильтра Блума для высокопроизводительного чтения секвенирования» . Биоинформатика . 30 (10): 1354–1362. doi : 10.1093/биоинформатика/btu030 . ISSN 1367-4803 . ПМК 6365934 . ПМИД 24451628 .
- ^ Пеллоу, Дэвид; Филиппова, Дарья; Кингсфорд, Карл (01 июня 2017 г.). «Улучшение производительности фильтра Блума для данных последовательностей с использованием фильтров Блума k-mer» . Журнал вычислительной биологии . 24 (6): 547–557. дои : 10.1089/cmb.2016.0155 . ISSN 1066-5277 . ПМК 5467106 . ПМИД 27828710 .
- ^ Чжан, Чжаоцзюнь; Ван, Вэй (15 июня 2014 г.). «RNA-Skim: быстрый метод количественного определения RNA-Seq на уровне транскриптов» . Биоинформатика . 30 (12): i283–i292. doi : 10.1093/биоинформатика/btu288 . ISSN 1367-4803 . ПМК 4058932 . ПМИД 24931995 .