Метагеномика
| Часть серии о |
| Генетика |
|---|
 |

Метагеномика — это исследование генетического материала, полученного непосредственно из экологических или клинических образцов с помощью метода, называемого секвенированием . Эту широкую область можно также назвать геномикой окружающей среды , экогеномикой , геномикой сообщества или микробиомикой .
В то время как традиционная микробиология микробных , секвенирование и геномика геномов полагаются на культивируемые клональные культуры , раннее секвенирование генов окружающей среды клонировало определенные гены (часто ген 16S рРНК ) для создания профиля разнообразия в природном образце. Такая работа показала, что подавляющее большинство микробного биоразнообразия было упущено из-за методов культивирования. [2]
Благодаря своей способности раскрывать ранее скрытое разнообразие микроскопической жизни, метагеномика предлагает мощный способ понимания микробного мира, который может революционизировать понимание биологии. [3] Поскольку стоимость секвенирования ДНК продолжает падать, метагеномика теперь позволяет экологию микробов исследовать в гораздо большем масштабе и деталях, чем раньше. В недавних исследованиях используется либо « дробовик », либо ПЦР- направленное секвенирование, чтобы получить в значительной степени объективные образцы всех генов от всех членов отобранных сообществ. [4]
Этимология [ править ]
Термин «метагеномика» впервые был использован Джо Хэндельсманом , Робертом М. Гудманом , Мишель Р. Рондон, Джоном Кларди и Шоном Ф. Брэйди и впервые появился в публикации в 1998 году. [5] Термин «метагеном» отсылал к идее о том, что совокупность генов, секвенированных из окружающей среды, можно анализировать аналогично изучению одного генома . В 2005 году Кевин Чен и Лиор Пахтер (исследователи из Калифорнийского университета в Беркли ) определили метагеномику как «применение современной техники геномики без необходимости изоляции и лабораторного культивирования отдельных видов». [6]
История [ править ]
| Часть серии о |
| ДНК-штрихкодирование |
|---|
 |
| По таксонам |
| Другой |
Традиционное секвенирование начинается с культивирования идентичных клеток в качестве источника ДНК . Однако ранние метагеномные исследования показали, что во многих средах, вероятно, существуют большие группы микроорганизмов, которые невозможно культивировать и, следовательно, невозможно секвенировать. Эти ранние исследования были сосредоточены на последовательностях 16S рибосомальной РНК (рРНК), которые относительно короткие, часто консервативны внутри вида и, как правило, различаются между видами. множество последовательностей 16S рРНК Было обнаружено , которые не принадлежат ни одному из известных культивируемых видов , что указывает на существование множества неизолированных организмов. Эти исследования генов рибосомальной РНК, взятых непосредственно из окружающей среды, показали, что методы, основанные на культивировании, менее 1% видов бактерий и архей . обнаруживают в образце [2] Большая часть интереса к метагеномике связана с этими открытиями, которые показали, что подавляющее большинство микроорганизмов ранее оставалось незамеченным.
В 1980-х годах первые молекулярные работы в этой области были проведены Норманом Р. Пейсом и его коллегами, которые использовали ПЦР для изучения разнообразия последовательностей рибосомальных РНК. [7] Результаты этих революционных исследований побудили Пейса еще в 1985 году предложить идею клонирования ДНК непосредственно из образцов окружающей среды. [8] Это привело к первому отчету об выделении и клонировании массива ДНК из образца окружающей среды, опубликованному Пейсом и его коллегами в 1991 году. [9] в то время как Пейс работал на факультете биологии в Университете Индианы . Значительные усилия гарантировали, что это не были ложноположительные результаты ПЦР , и подтвердили существование сложного сообщества неисследованных видов. Хотя эта методология ограничивалась исследованием высококонсервативных, некодирующих белки генов , она подтвердила ранние наблюдения, основанные на морфологии микробов, о том, что разнообразие было гораздо более сложным, чем было известно методами культивирования. Вскоре после этого, в 1995 году, Хили сообщил о метагеномном выделении функциональных генов из «зообиблиотек», сконструированных из сложной культуры организмов окружающей среды, выращенных в лаборатории на сушеных травах . [10] Покинув лабораторию Пейса, Эдвард ДеЛонг продолжил работу в этой области и опубликовал работу, которая в значительной степени заложила основу для филогении окружающей среды, основанной на характерных последовательностях 16S, начиная с создания его группой библиотек из морских образцов. [11]
В 2002 году Майя Брейтбарт , Форест Ровер и их коллеги использовали метод секвенирования окружающей среды (см. ниже), чтобы показать, что 200 литров морской воды содержат более 5000 различных вирусов. [12] Последующие исследования показали, что в фекалиях человека содержится более тысячи видов вирусов и, возможно, миллион различных вирусов на килограмм морских отложений , включая множество бактериофагов . По сути, все вирусы в этих исследованиях представляли собой новые виды. В 2004 году Джин Тайсон, Джилл Бэнфилд и их коллеги из Калифорнийского университета в Беркли и Объединенного института генома секвенировали ДНК, извлеченную из дренажной системы кислотной шахты . [13] В результате этих усилий были получены полные или почти полные геномы горстки бактерий и архей , которые ранее сопротивлялись попыткам их культивирования. [14]
Начиная с 2003 года Крейг Вентер , руководитель параллельного проекта «Геном человека» , финансируемого из частных источников, возглавил Глобальную экспедицию по отбору проб океана (GOS), совершая кругосветное плавание и собирая метагеномные образцы на протяжении всего путешествия. Все эти образцы были секвенированы с использованием дробовика в надежде, что будут идентифицированы новые геномы (и, следовательно, новые организмы). Пилотный проект, проведенный в Саргассовом море , обнаружил ДНК почти 2000 различных видов , включая 148 типов бактерий, никогда ранее не встречавшихся. [15] Вентер тщательно исследовал западное побережье Соединенных Штатов и в 2006 году завершил двухлетнюю экспедицию по исследованию Балтийского , Средиземного и Черного морей. Анализ метагеномных данных, собранных во время этого путешествия, выявил две группы организмов: одна состоит из таксонов, адаптированных к условиям окружающей среды «пир или голод», а вторая состоит из относительно меньшего количества, но более обильных и широко распространенных таксонов, состоящих в основном из планктона . [16]
В 2005 году Стефан Шустер из Университета штата Пенсильвания и его коллеги опубликовали первые последовательности образца окружающей среды, полученные с помощью высокопроизводительного секвенирования , в данном случае массово-параллельного пиросеквенирования, разработанного 454 Life Sciences . [17] Еще одна ранняя статья в этой области появилась в 2006 году Робертом Эдвардсом, Форестом Ровером и коллегами из Государственного университета Сан-Диего . [18]
Секвенирование [ править ]

Восстановление последовательностей ДНК длиной более нескольких тысяч пар оснований из образцов окружающей среды было очень трудным, пока недавние достижения в области молекулярно-биологических методов не позволили создать библиотеки в бактериальных искусственных хромосомах (BAC), которые предоставили лучшие векторы для молекулярного клонирования . [20]
дробовика Метагеномика
Достижения в области биоинформатики , совершенствование амплификации ДНК и распространение вычислительных мощностей во многом помогли анализу последовательностей ДНК, извлеченных из образцов окружающей среды, что позволило адаптировать дробовое секвенирование к метагеномным образцам (известное также как дробовое секвенирование полного метагенома или секвенирование WMGS). Этот подход, используемый для секвенирования многих культивируемых микроорганизмов и генома человека , случайным образом разрезает ДНК, секвенирует множество коротких последовательностей и реконструирует их в консенсусную последовательность . Секвенирование методом дробовика выявляет гены, присутствующие в образцах окружающей среды. Исторически для облегчения такого секвенирования использовались библиотеки клонов. Однако с развитием технологий высокопроизводительного секвенирования этап клонирования больше не нужен, и больший выход данных секвенирования можно получить без этого трудоемкого этапа. Метагеномика дробовика предоставляет информацию как о том, какие организмы присутствуют, так и о том, какие метаболические процессы возможны в сообществе. [21] Поскольку сбор ДНК из окружающей среды в значительной степени не контролируется, наиболее распространенные организмы в образце окружающей среды наиболее широко представлены в результирующих данных о последовательностях. Для достижения высокого охвата, необходимого для полного определения геномов недостаточно представленных членов сообщества, необходимы большие выборки, часто непомерно высокие. С другой стороны, случайный характер дробового секвенирования гарантирует, что многие из этих организмов, которые в противном случае остались бы незамеченными при использовании традиционных методов культивирования, будут представлены по крайней мере несколькими небольшими сегментами последовательности. [13]
Высокопроизводительное секвенирование
Преимущество высокопроизводительного секвенирования заключается в том, что этот метод не требует клонирования ДНК перед секвенированием, что устраняет одну из основных ошибок и узких мест при отборе проб окружающей среды. Первые метагеномные исследования, проведенные с использованием высокопроизводительного секвенирования, использовали массовое параллельное пиросеквенирование 454 . [17] Три другие технологии, обычно применяемые для отбора проб окружающей среды, — это персональная геномная машина Ion Torrent , Illumina MiSeq или HiSeq и система SOLiD компании Applied Biosystems . [22] Эти методы секвенирования ДНК позволяют получить более короткие фрагменты, чем секвенирование Сэнгера ; Система Ion Torrent PGM и пиросеквенирование 454 обычно производят считывания размером ~400 п.н., Illumina MiSeq производит считывания 400–700 п.н. (в зависимости от того, используются ли варианты с парными концами), а SOLiD производит считывания 25–75 п.н. [23] Исторически сложилось так, что эти длины считывания были значительно короче, чем типичная длина считывания секвенирования Сэнгера, составляющая ~ 750 п.о., однако технология Illumina быстро приближается к этому эталону. Однако это ограничение компенсируется гораздо большим количеством считываний последовательности. В 2009 году пиросеквенированные метагеномы генерировали 200–500 мегабаз, а платформы Illumina генерировали около 20–50 гигабаз, но в последние годы эти результаты увеличились на порядки. [24]
Новый подход сочетает в себе дробовое секвенирование и захват конформации хромосом (Hi-C), который измеряет близость любых двух последовательностей ДНК в одной и той же клетке, чтобы управлять сборкой микробного генома. [25] Технологии секвенирования длинных считываний, в том числе PacBio RSII и PacBio Sequel от Pacific Biosciences , а также Nanopore MinION, GridION, PromethION от Oxford Nanopore Technologies , являются еще одним выбором для получения длинных считываний дробового секвенирования, которые должны облегчить процесс сборки. [26]
Биоинформатика [ править ]
В этом разделе отсутствует информация об оценке качества: по сборке (N50, MetaQUAST), по геному (универсальные однокопийные маркерные гены – CheckM и BUSCO). ( февраль 2022 г. ) |
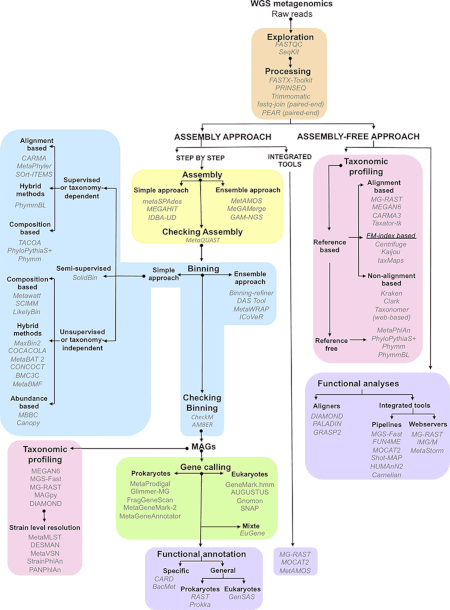
Данные, полученные в ходе экспериментов по метагеномике, огромны и по своей сути зашумлены: они содержат фрагментированные данные, представляющие до 10 000 видов. [1] Секвенирование метагенома рубца коровы позволило получить 279 гигабаз , или 279 миллиардов пар оснований данных нуклеотидной последовательности. [28] кишечника человека в то время как каталог генов микробиома идентифицировал 3,3 миллиона генов, собранных из 567,7 гигабаз данных о последовательностях. [29] Сбор, обработка и извлечение полезной биологической информации из наборов данных такого размера представляют собой серьезные вычислительные проблемы для исследователей. [21] [30] [31] [32]
Предварительная фильтрация последовательности [ править ]
Первый этап анализа метагеномных данных требует выполнения определенных этапов предварительной фильтрации, включая удаление избыточных последовательностей низкого качества и последовательностей вероятного эукариотического происхождения (особенно в метагеномах человеческого происхождения). [33] [34] Доступные методы удаления загрязняющих последовательностей геномной ДНК эукариот включают Eu-Detect и DeConseq. [35] [36]
Сборка [ править ]
Данные о последовательностях ДНК из геномных и метагеномных проектов по существу одинаковы, но данные о геномных последовательностях обеспечивают более широкий охват , в то время как метагеномные данные обычно не являются избыточными. [31] Более того, более широкое использование технологий секвенирования второго поколения с короткой длиной считывания означает, что большая часть будущих метагеномных данных будет подвержена ошибкам. В совокупности эти факторы делают сборку считывания метагеномных последовательностей в геномах сложной и ненадежной. Неправильная сборка вызвана наличием повторяющихся последовательностей ДНК , которые особенно затрудняют сборку из-за разницы в относительном обилии видов, присутствующих в образце. [37] Неправильная сборка может также включать объединение последовательностей более чем одного вида в химерные контиги . [37]
Существует несколько программ сборки, большинство из которых могут использовать информацию из парных тегов для повышения точности сборки. Некоторые программы, такие как Phrap или Celera Assembler, были разработаны для сборки отдельных геномов , но, тем не менее, дают хорошие результаты при сборке наборов метагеномных данных. [1] Другие программы, такие как ассемблер Velvet , были оптимизированы для более коротких чтений, производимых секвенированием второго поколения с использованием графов де Брейна . [38] [39] Использование эталонных геномов позволяет исследователям улучшить сборку наиболее распространенных видов микробов, но этот подход ограничен небольшим подмножеством типов микробов, для которых доступны секвенированные геномы. [37] После создания сборки дополнительной задачей является «метагеномная деконволюция» или определение того, какие последовательности принадлежат каким видам в образце. [40]
Генное предсказание [ править ]
метагеномного анализа Конвейеры используют два подхода к аннотации кодирующих областей в собранных контигах. [37] Первый подход заключается в идентификации генов на основе гомологии с генами, которые уже общедоступны в базах данных последовательностей , обычно с помощью BLAST поиска . Такой подход реализован в программе МЕГАН 4. [41] Второй, ab initio , использует внутренние особенности последовательности для прогнозирования кодирующих областей на основе обучающих наборов генов родственных организмов. Именно такой подход используют такие программы, как GeneMark. [42] и ГЛИММЕР . Основное преимущество предсказания ab initio заключается в том, что оно позволяет обнаруживать кодирующие области, у которых отсутствуют гомологи в базах данных последовательностей; однако он наиболее точен, когда для сравнения доступны большие участки смежной геномной ДНК. [1]
Видовое разнообразие [ править ]
Генные аннотации дают ответ «что», а измерения видового разнообразия дают ответ «кто». [43] Чтобы связать состав сообщества и функции в метагеномах, последовательности необходимо объединить. Биннинг — это процесс связывания определенной последовательности с организмом. [37] При объединении на основе сходства такие методы, как BLAST, используются для быстрого поиска филогенетических маркеров или других подобных последовательностей в существующих общедоступных базах данных. Такой подход реализован в МЕГАН . [44] Другой инструмент, PhymmBL, использует интерполированные марковские модели для назначения показаний. [1] MetaPhlAn и AMPHORA — это методы, основанные на уникальных маркерах, специфичных для клады, для оценки относительной численности организмов с улучшенными вычислительными характеристиками. [45] Другие инструменты, такие как mOTU [46] [47] и МетаФилер, [48] использовать универсальные маркерные гены для профилирования видов прокариот. С помощью профилировщика mOTUs можно составлять профили видов без эталонного генома, что улучшает оценку разнообразия микробного сообщества. [47] Последние методы, такие как SLIMM , используют ландшафт покрытия чтения отдельных эталонных геномов, чтобы минимизировать ложноположительные совпадения и получить надежную относительную численность. [49] При биннинге на основе композиции методы используют внутренние особенности последовательности, такие как частоты олигонуклеотидов или систематическая ошибка использования кодонов . [1] После группирования последовательностей можно провести сравнительный анализ разнообразия и богатства.
Интеграция данных [ править ]
Огромный объем экспоненциально растущих данных о последовательностях представляет собой сложную задачу, которая осложняется сложностью метаданных, связанных с метагеномными проектами. Метаданные включают подробную информацию о трехмерной (включая глубину и высоту) географии и характеристиках окружающей среды образца, физических данных о месте отбора проб и методологии отбора проб. [31] Эта информация необходима как для обеспечения воспроизводимости , так и для возможности последующего анализа. Из-за своей важности метаданные, а также совместный обзор и обработка данных требуют стандартизированных форматов данных, расположенных в специализированных базах данных, таких как база данных Genomes OnLine Database (GOLD). [50]
Было разработано несколько инструментов для интеграции метаданных и данных о последовательностях, что позволяет проводить последующий сравнительный анализ различных наборов данных с использованием ряда экологических индексов. В 2007 году Фолкер Мейер и Роберт Эдвардс и команда Аргоннской национальной лаборатории и Чикагского университета выпустили «Быструю аннотацию метагеномики с использованием сервера Subsystem Technology» ( MG-RAST ) — ресурс сообщества для анализа наборов метагеномных данных. [51] По состоянию на июнь 2012 г. более 14,8 терабаз (14x10 12 баз) ДНК были проанализированы, при этом более 10 000 общедоступных наборов данных свободно доступны для сравнения в рамках MG-RAST. В настоящее время более 8000 пользователей отправили в MG-RAST в общей сложности 50 000 метагеномов. Система «Интегрированные микробные геномы/метагеномы » (IMG/M) также предоставляет набор инструментов для функционального анализа микробных сообществ на основе их метагеномной последовательности, основанной на эталонных геномах изолятов, включенных в систему «Интегрированные микробные геномы » (IMG) и Геномную энциклопедию Проект «Бактерии и археи» (GEBA) . [52]
Одним из первых автономных инструментов для высокопроизводительного анализа метагеномных данных был MEGAN (MEta Genome ANalyzer). [41] [44] Первая версия программы была использована в 2005 году для анализа метагеномного контекста последовательностей ДНК, полученных из кости мамонта. [17] На основе сравнения BLAST со справочной базой данных этот инструмент выполняет как таксономическое, так и функциональное группирование, помещая чтения в узлы таксономии NCBI с использованием простого алгоритма наименьшего общего предка (LCA) или в узлы SEED или KEGG. классификаций , соответственно. [53]
С появлением быстрых и недорогих инструментов для секвенирования рост баз данных последовательностей ДНК стал экспоненциальным (например, база данных NCBI GenBank). [54] ). Чтобы идти в ногу с высокопроизводительным секвенированием, необходимы более быстрые и эффективные инструменты, поскольку подходы на основе BLAST, такие как MG-RAST или MEGAN, работают медленно для аннотирования больших образцов (например, несколько часов для обработки набора данных/образца небольшого/среднего размера). [55] ). Так, в последнее время появились сверхбыстрые классификаторы благодаря более доступным мощным серверам. Эти инструменты могут выполнять таксономическую аннотацию с чрезвычайно высокой скоростью, например CLARK. [56] (по словам авторов CLARK, он может точно классифицировать «32 миллиона метагеномных коротких чтений в минуту»). При такой скорости очень большой набор данных/выборка из миллиарда коротких чтений может быть обработан примерно за 30 минут.
С увеличением доступности образцов, содержащих древнюю ДНК, и из-за неопределенности, связанной с природой этих образцов (повреждение древней ДНК), [57] стал доступен быстрый инструмент, способный производить консервативные оценки сходства. По словам авторов FALCON, он может использовать смягченные пороги и редактировать расстояния, не влияя на память и скорость работы.
метагеномика Сравнительная
Сравнительный анализ метагеномов может дать дополнительное представление о функции сложных микробных сообществ и их роли в здоровье хозяина. [58] Парные или множественные сравнения метагеномов могут проводиться на уровне состава последовательностей (сравнение содержания GC или размера генома), таксономического разнообразия или функционального комплемента. Сравнение популяционной структуры и филогенетического разнообразия можно проводить на основе 16S рРНК и других генов-филогенетических маркеров или — в случае сообществ с низким разнообразием — путем реконструкции генома по набору метагеномных данных. [59] Функциональные сравнения между метагеномами могут быть выполнены путем сравнения последовательностей со справочными базами данных, такими как COG или KEGG , а также составления таблицы распространенности по категориям и оценки любых различий на предмет статистической значимости. [53] Этот геноцентричный подход подчеркивает функциональный состав сообщества в целом, а не таксономических групп, и показывает, что функциональные дополнения аналогичны в аналогичных условиях окружающей среды. [59] Следовательно, метаданные об экологическом контексте метагеномной выборки особенно важны в сравнительном анализе, поскольку они дают исследователям возможность изучать влияние среды обитания на структуру и функционирование сообщества. [1]
Кроме того, в нескольких исследованиях также использовались модели использования олигонуклеотидов для выявления различий между различными микробными сообществами. Примеры таких методологий включают подход относительного содержания динуклеотидов Willner et al. [60] и подход HabiSign Ghosh et al. [61] Это последнее исследование также показало, что различия в моделях использования тетрануклеотидов могут быть использованы для идентификации генов (или метагеномных чтений), происходящих из конкретных сред обитания. Кроме того, некоторые методы, такие как TriageTools [62] или Сравнить объявления [63] обнаруживать аналогичные чтения между двумя наборами чтения. Мера сходства, которую они применяют при чтении, основана на количестве идентичных слов длины k, общих для пар чтений.
Ключевой целью сравнительной метагеномики является идентификация микробных групп, которые отвечают за придание определенных характеристик данной среде. Однако из-за проблем в технологиях секвенирования необходимо учитывать артефакты, как в MetagenomeSeq. [30] Другие охарактеризовали межмикробные взаимодействия между резидентными микробными группами. Приложение сравнительного метагеномного анализа на основе графического пользовательского интерфейса под названием Community-Analyzer было разработано Kuntal et al. [64] который реализует алгоритм компоновки графов на основе корреляции, который не только облегчает быструю визуализацию различий в анализируемых микробных сообществах (с точки зрения их таксономического состава), но также дает представление о присущих им межмикробных взаимодействиях, происходящих в них. Примечательно, что этот алгоритм компоновки также позволяет группировать метагеномы на основе вероятных моделей межмикробного взаимодействия, а не просто сравнивать значения численности различных таксономических групп. Кроме того, инструмент реализует несколько интерактивных функций на основе графического пользовательского интерфейса, которые позволяют пользователям выполнять стандартный сравнительный анализ микробиомов.
Анализ данных [ править ]
Метаболизм сообщества [ править ]
Во многих бактериальных сообществах, природных или искусственно созданных (например, в биореакторах ), существует значительное разделение труда в метаболизме ( синтрофия ), в ходе которого продукты жизнедеятельности одних организмов являются метаболитами для других. [65] В одной из таких систем, метаногенном биореакторе, функциональная стабильность требует присутствия нескольких синтрофных видов ( Syntropobacterales и Synergistia ), работающих вместе, чтобы превратить сырьевые ресурсы в полностью метаболизированные отходы ( метан ). [66] Используя сравнительные исследования генов и эксперименты по экспрессии с использованием микрочипов или протеомики, исследователи могут собрать воедино метаболическую сеть, выходящую за рамки видовых границ. Такие исследования требуют детальных знаний о том, какие версии каких белков кодируются какими видами и даже какими штаммами каких видов. Таким образом, геномная информация сообщества является еще одним фундаментальным инструментом (наряду с метаболомикой и протеомикой) в поисках определения того, как метаболиты передаются и трансформируются сообществом. [67]
Метатранскриптомика [ править ]
Метагеномика позволяет исследователям получить доступ к функциональному и метаболическому разнообразию микробных сообществ, но не может показать, какие из этих процессов активны. [59] Извлечение и анализ метагеномной мРНК ( метатранскриптома ) предоставляет информацию о профилях регуляции и экспрессии сложных сообществ. Из-за технических трудностей ( например, короткого периода полураспада мРНК) при сборе РНК из окружающей среды на сегодняшний день было проведено относительно мало in situ . метатранскриптомных исследований микробных сообществ [59] Хотя первоначально исследования метатранскриптомики ограничивались технологией микрочипов , в них использовались технологии транскриптомики для измерения полногеномной экспрессии и количественной оценки микробного сообщества. [59] впервые был использован для анализа окисления аммиака в почвах. [68]
Вирусы [ править ]
Метагеномное секвенирование особенно полезно при изучении вирусных сообществ. Поскольку у вирусов нет общего универсального филогенетического маркера (например, 16S РНК для бактерий и архей и 18S РНК для эукарий), единственный способ получить доступ к генетическому разнообразию вирусного сообщества из образца окружающей среды — это метагеномика. Таким образом, вирусные метагеномы (также называемые виромами) должны предоставлять все больше и больше информации о вирусном разнообразии и эволюции. [69] [70] [71] [72] [73] Например, метагеномный конвейер под названием Giant Virus Finder показал первые доказательства существования гигантских вирусов в солончаковой пустыне. [74] и в сухих долинах Антарктики. [75]
Приложения [ править ]
Метагеномика обладает потенциалом для развития знаний в самых разных областях. Его также можно применять для решения практических задач в медицине , инженерии , сельском хозяйстве , устойчивом развитии и экологии . [31] [76]
Сельское хозяйство [ править ]
Почвы , на которых произрастают растения, населены микробными сообществами: в одном грамме почвы содержится около 10 9 -10 10 микробные клетки, которые содержат около одной гигабазы информации о последовательностях. [77] [78] Микробные сообщества, населяющие почвы, являются одними из самых сложных, известных науке, и остаются плохо изученными, несмотря на их экономическое значение. [79] Микробные консорциумы выполняют широкий спектр экосистемных услуг , необходимых для роста растений, включая фиксацию атмосферного азота , круговорот питательных веществ , подавление болезней и связывание железа и других металлов . [80] Стратегии функциональной метагеномики используются для изучения взаимодействия между растениями и микробами посредством независимого от культивирования изучения этих микробных сообществ. [81] [82] Позволяя понять роль ранее некультивируемых или редких членов сообщества в круговороте питательных веществ и стимулировании роста растений, метагеномные подходы могут способствовать улучшению выявления болезней сельскохозяйственных культур и домашнего скота , а также адаптации улучшенных методов ведения сельского хозяйства , которые улучшают здоровье сельскохозяйственных культур, используя взаимосвязь. между микробами и растениями. [31]
Биотопливо [ править ]
Биотопливо — это топливо, полученное в результате переработки биомассы , например, при преобразовании целлюлозы , содержащейся в кукурузы стеблях , просеяном и другой биомассе, в целлюлозный этанол . [31] Этот процесс зависит от микробных консорциумов (ассоциаций), которые преобразуют целлюлозу в сахара с последующей ферментацией сахаров в этанол . Микробы также производят различные источники биоэнергии, включая метан и водород . [31]
Эффективное в промышленных масштабах разрушение биомассы требует новых ферментов с более высокой производительностью и меньшей стоимостью. [28] Метагеномные подходы к анализу сложных микробных сообществ позволяют проводить целенаправленный скрининг ферментов, имеющих промышленное применение в производстве биотоплива, таких как гликозидгидролазы . [83] Более того, для контроля над ними необходимы знания о том, как функционируют эти микробные сообщества, а метагеномика является ключевым инструментом в их понимании. Метагеномные подходы позволяют проводить сравнительный анализ между конвергентными микробными системами, такими как биогаза. ферментеры [84] или насекомые травоядные , такие как грибной сад муравьев -листорезов . [85]
Биотехнология [ править ]
Микробные сообщества производят широкий спектр биологически активных химических веществ, которые используются в конкуренции и общении. [80] Многие из используемых сегодня лекарств изначально были обнаружены в микробах; недавний прогресс в разработке богатого генетического ресурса некультивируемых микробов привел к открытию новых генов, ферментов и натуральных продуктов. [59] [86] Применение метагеномики позволило разработать товарные и тонкие химикаты , агрохимикаты и фармацевтические препараты преимущество катализируемого ферментами . хирального синтеза, , где все больше признается [87]
метагеномных данных используются два типа анализа При биоразведке : функциональный скрининг выраженного признака и скрининг на основе последовательности интересующих последовательностей ДНК. [88] Функциональный анализ направлен на идентификацию клонов, проявляющих желаемый признак или полезную активность, с последующей биохимической характеристикой и анализом последовательности. Этот подход ограничен наличием подходящего скрининга и требованием экспрессии желаемого признака в клетке-хозяине. Более того, низкий уровень обнаружения (менее одного на 1000 проверенных клонов) и трудоемкий характер еще больше ограничивают этот подход. [89] Напротив, анализ, основанный на последовательностях, использует консервативные последовательности ДНК для разработки праймеров для ПЦР для скрининга клонов на предмет интересующей последовательности. [88] По сравнению с подходами, основанными на клонировании, использование подхода, основанного только на последовательностях, еще больше снижает объем необходимой лабораторной работы. Применение массово-параллельного секвенирования также значительно увеличивает объем генерируемых данных о последовательностях, что требует высокопроизводительных конвейеров биоинформатического анализа. [89] Подход к скринингу, основанный на последовательностях, ограничен широтой и точностью функций генов, представленных в общедоступных базах данных последовательностей. На практике в экспериментах используется комбинация функционального и последовательностного подходов, основанная на интересующей функции, сложности проверяемого образца и других факторах. [89] [90] Примером успеха использования метагеномики в качестве биотехнологии для открытия лекарств является антибиотик малацидин . [91]
Экология [ править ]

Метагеномика может дать ценную информацию о функциональной экологии экологических сообществ. [92] Метагеномный анализ бактериальных консорциумов, обнаруженных в испражнениях австралийских морских львов, предполагает, что богатые питательными веществами фекалии морских львов могут быть важным источником питательных веществ для прибрежных экосистем. Это связано с тем, что бактерии, которые выделяются одновременно с дефекацией, способны расщеплять питательные вещества, содержащиеся в фекалиях, в биодоступную форму, которая может быть использована в пищевой цепи. [93]
Секвенирование ДНК также можно использовать в более широком смысле для идентификации видов, присутствующих в водоеме. [94] отфильтрованный из воздуха мусор, образцы грязи или фекалий животных, [95] и даже обнаруживать продукты питания по еде с кровью. [96] Это позволит установить ареал инвазивных видов и видов, находящихся под угрозой исчезновения , а также отслеживать сезонные популяции.
Восстановление окружающей среды
Метагеномика может улучшить стратегии мониторинга воздействия загрязнителей на экосистемы и очистки загрязненной окружающей среды. Более глубокое понимание того, как микробные сообщества справляются с загрязнителями, улучшает оценку потенциала загрязненных территорий для восстановления после загрязнения и увеличивает шансы биоаугментации или биостимуляции . на успех испытаний [97]
кишечных Характеристика микробов
Микробные сообщества играют ключевую роль в сохранении здоровья человека , но их состав и механизм, с помощью которого они это делают, остаются загадкой. [98] Метагеномное секвенирование используется для характеристики микробных сообществ из 15–18 участков тела как минимум 250 человек. Это часть инициативы «Микробиом человека», основная цель которой — определить, существует ли основной микробиом человека , понять изменения в микробиоме человека, которые можно коррелировать со здоровьем человека, а также разработать новые технологические и биоинформатические инструменты для поддержки этих целей. [99]
В другом медицинском исследовании в рамках проекта MetaHit (Метагеномика кишечного тракта человека) приняли участие 124 человека из Дании и Испании, среди которых были здоровые пациенты с избыточным весом и раздраженным кишечником. [100] В исследовании была предпринята попытка классифицировать глубину и филогенетическое разнообразие желудочно-кишечных бактерий. Используя данные последовательности Illumina GA и SOAPdenovo, инструмент на основе графов де Брейна, специально разработанный для коротких чтений сборки, они смогли сгенерировать 6,58 миллиона контигов размером более 500 п.н. с общей длиной контига 10,3 Гб и длиной N50 2,2 КБ.
Исследование показало, что два бактериальных подразделения, Bacteroidetes и Firmicutes, составляют более 90% известных филогенетических категорий, которые доминируют в бактериях дистальных отделов кишечника. Используя относительные частоты генов, обнаруженных в кишечнике, исследователи определили 1244 метагеномных кластера, которые критически важны для здоровья кишечного тракта. В этих кластерах диапазонов выделяются два типа функций: хозяйственные и специфичные для кишечника. Кластеры генов «домашнего хозяйства» необходимы всем бактериям и часто играют важную роль в основных метаболических путях, включая центральный метаболизм углерода и синтез аминокислот. Специфические для кишечника функции включают адгезию к белкам хозяина и сбор сахаров из гликолипидов шаровидного ряда. Было показано, что у пациентов с синдромом раздраженного кишечника наблюдается на 25% меньше генов и меньшее бактериальное разнообразие, чем у людей, не страдающих синдромом раздраженного кишечника, что указывает на то, что изменения в разнообразии биома кишечника пациентов могут быть связаны с этим заболеванием. [100]
Хотя эти исследования подчеркивают некоторые потенциально ценные медицинские применения, только 31–48,8% чтений могут быть сопоставлены со 194 общедоступными бактериальными геномами кишечника человека и 7,6–21,2% с бактериальными геномами, доступными в GenBank, что указывает на то, что необходимо еще гораздо больше исследований, чтобы захватывать новые бактериальные геномы. [101]
В рамках проекта «Микробиом человека » (HMP) микробные сообщества кишечника анализировались с использованием высокопроизводительного секвенирования ДНК. HMP показал, что, в отличие от отдельных видов микробов, многие метаболические процессы присутствуют во всех средах обитания организма с различной частотой. В рамках проекта микробиома человека были изучены микробные сообщества 649 метагеномов, взятых из семи первичных участков тела 102 человек . Метагеномный анализ выявил различия в специфическом количестве ниш среди 168 функциональных модулей и 196 метаболических путей внутри микробиома. Они включали деградацию гликозаминогликанов в кишечнике, а также транспорт фосфатов и аминокислот, связанный с фенотипом хозяина (рН влагалища) в заднем своде. HMP выявил полезность метагеномики в диагностике и доказательной медицине . Таким образом, метагеномика является мощным инструментом для решения многих актуальных проблем в области персонализированной медицины . [102]
У животных метагеномика может использоваться для составления профиля микробиома кишечника и обнаружения бактерий, устойчивых к антибиотикам. [103] Это может иметь значение для мониторинга распространения болезней от диких животных к сельскохозяйственным животным и людям.
Диагностика инфекционных заболеваний [ править ]
Дифференциация инфекционных и неинфекционных заболеваний и выявление основной этиологии инфекции может быть сложной задачей. Например, более половины случаев энцефалита остаются недиагностированными, несмотря на обширное тестирование с использованием самых современных клинических лабораторных методов. Клиническое метагеномное секвенирование перспективно в качестве чувствительного и быстрого метода диагностики инфекции путем сравнения генетического материала, обнаруженного в образце пациента, с базами данных всех известных микроскопических патогенов человека и тысяч других бактериальных, вирусных, грибковых и паразитических организмов, а также базами данных по генам устойчивости к противомикробным препаратам. последовательности с соответствующими клиническими фенотипами. [104]
Наблюдение арбовирусами за
Метагеномика стала бесценным инструментом, помогающим охарактеризовать разнообразие и экологию патогенов, переносимых гематофагами (кровопитающимися) насекомыми, такими как комары и клещи. [105] [106] [107] Метагеномика – это [ когда? ] обычно используется должностными лицами и организациями общественного здравоохранения [ где? ] для надзора за арбовирусами . [108] [109]
См. также [ править ]
Ссылки [ править ]
- ^ Jump up to: Перейти обратно: а б с д и ж г Вули Дж. К., Годзик А., Фридберг I (февраль 2010 г.). Борн PE (ред.). «Букварь по метагеномике» . PLOS Вычислительная биология . 6 (2): e1000667. Бибкод : 2010PLSCB...6E0667W . дои : 10.1371/journal.pcbi.1000667 . ПМК 2829047 . ПМИД 20195499 .
- ^ Jump up to: Перейти обратно: а б Хугенгольц П., Гебель Б.М., Пейс Н.Р. (сентябрь 1998 г.). «Влияние независимых от культуры исследований на формирующийся филогенетический взгляд на бактериальное разнообразие» . Журнал бактериологии . 180 (18): 4765–74. дои : 10.1128/JB.180.18.4765-4774.1998 . ПМЦ 107498 . ПМИД 9733676 .
- ^ Марко, Д., изд. (2011). Метагеномика: текущие инновации и будущие тенденции . Кайстер Академик Пресс . ISBN 978-1-904455-87-5 .
- ^ Эйзен Дж. А. (март 2007 г.). «Экологическое секвенирование дробовика: его потенциал и проблемы для изучения скрытого мира микробов» . ПЛОС Биология . 5 (3): е82. doi : 10.1371/journal.pbio.0050082 . ПМК 1821061 . ПМИД 17355177 .
- ^ Хэндельсман Дж., Рондон М.Р., Брэди С.Ф., Кларди Дж., Гудман Р.М. (октябрь 1998 г.). «Молекулярно-биологический доступ к химии неизвестных почвенных микробов: новый рубеж для натуральных продуктов» . Химия и биология . 5 (10): Р245-9. дои : 10.1016/S1074-5521(98)90108-9 . ПМИД 9818143 . .
- ^ Чен К., Пахтер Л. (июль 2005 г.). «Биоинформатика для полногеномного секвенирования микробных сообществ» . PLOS Вычислительная биология . 1 (2): 106–12. Бибкод : 2005PLSCB...1...24C . дои : 10.1371/journal.pcbi.0010024 . ПМЦ 1185649 . ПМИД 16110337 .
- ^ Лейн DJ, Пейс Б., Олсен Г.Дж., Шталь Д.А., Согин М.Л., Пейс Н.Р. (октябрь 1985 г.). «Быстрое определение последовательностей 16S рибосомальной РНК для филогенетического анализа» . Труды Национальной академии наук Соединенных Штатов Америки . 82 (20): 6955–9. Бибкод : 1985PNAS...82.6955L . дои : 10.1073/pnas.82.20.6955 . ПМК 391288 . ПМИД 2413450 .
- ^ Пейс Н.Р., Шталь Д.А., Лейн DJ, Олсен Г.Дж. (1986). «Анализ природных микробных популяций по последовательностям рибосомальной РНК». В Маршалле К.С. (ред.). Достижения микробной экологии . Том. 9. Спрингер США. стр. 1–55. дои : 10.1007/978-1-4757-0611-6_1 . ISBN 978-1-4757-0611-6 .
- ^ Шмидт Т.М. , Делонг Э.Ф., Пейс Н.Р. (июль 1991 г.). «Анализ сообщества морского пикопланктона путем клонирования и секвенирования гена 16S рРНК» . Журнал бактериологии . 173 (14): 4371–8. дои : 10.1128/jb.173.14.4371-4378.1991 . ПМК 208098 . ПМИД 2066334 .
- ^ Хили Ф.Г., Рэй Р.М., Олдрич Х.К., Уилки А.С., Ингрэм Л.О., Шанмугам К.Т. (1995). «Прямое выделение функциональных генов, кодирующих целлюлазы, из микробных консорциумов в термофильном анаэробном варочном котле, поддерживаемом на лигноцеллюлозе». Прикладная микробиология и биотехнология . 43 (4): 667–74. дои : 10.1007/BF00164771 . ПМИД 7546604 . S2CID 31384119 .
- ^ Стейн Дж.Л., Марш Т.Л., Ву К.Ю., Шизуя Х., Делонг Э.Ф. (февраль 1996 г.). «Характеристика некультивируемых прокариот: выделение и анализ фрагмента генома длиной 40 тысяч пар оснований планктонного морского архея» . Журнал бактериологии . 178 (3): 591–9. дои : 10.1128/jb.178.3.591-599.1996 . ПМК 177699 . ПМИД 8550487 .
- ^ Брейтбарт М., Саламон П., Андресен Б., Махаффи Дж.М., Сигалл А.М., Мид Д. и др. (октябрь 2002 г.). «Геномный анализ некультивируемых морских вирусных сообществ» . Труды Национальной академии наук Соединенных Штатов Америки . 99 (22): 14250–5. Бибкод : 2002PNAS...9914250B . дои : 10.1073/pnas.202488399 . ПМК 137870 . ПМИД 12384570 .
- ^ Jump up to: Перейти обратно: а б Тайсон Г.В., Чепмен Дж., Хугенхольц П., Аллен Э.Э., Рам Р.Дж., Ричардсон П.М. и др. (март 2004 г.). «Структура сообщества и метаболизм посредством реконструкции микробных геномов из окружающей среды». Природа . 428 (6978): 37–43. Бибкод : 2004Natur.428...37T . дои : 10.1038/nature02340 . ПМИД 14961025 . S2CID 4420754 . (требуется подписка)
- ^ Гугенгольц П. (2002). «Изучение прокариотического разнообразия в эпоху генома» . Геномная биология . 3 (2): ОБЗОРЫ0003. doi : 10.1186/gb-2002-3-2-reviews0003 . ПМК 139013 . ПМИД 11864374 .
- ^ Вентер Дж.К., Ремингтон К., Гейдельберг Дж.Ф., Халперн А.Л., Раш Д., Эйзен Дж.А. и др. (апрель 2004 г.). «Дробовиковое секвенирование экологического генома Саргассова моря». Наука . 304 (5667): 66–74. Бибкод : 2004Sci...304...66V . CiteSeerX 10.1.1.124.1840 . дои : 10.1126/science.1093857 . ПМИД 15001713 . S2CID 1454587 .
- ^ Юсеф С., Нилсон К.Х., Раш Д.Б., МакКроу Дж.П., Дюпон К.Л., Ким М. и др. (ноябрь 2010 г.). «Геномная и функциональная адаптация планктонных прокариот поверхностного океана» . Природа . 468 (7320): 60–6. Бибкод : 2010Природа.468...60Г . дои : 10.1038/nature09530 . ПМИД 21048761 . (требуется подписка)
- ^ Jump up to: Перейти обратно: а б с Пойнар Х.Н., Шварц С., Ци Дж., Шапиро Б., Макфи Р.Д., Бюиг Б. и др. (январь 2006 г.). «От метагеномики до палеогеномики: крупномасштабное секвенирование ДНК мамонта». Наука . 311 (5759): 392–4. Бибкод : 2006Sci...311..392P . дои : 10.1126/science.1123360 . ПМИД 16368896 . S2CID 11238470 .
- ^ Эдвардс Р.А., Родригес-Брито Б., Уэгли Л., Хейнс М., Брейтбарт М., Петерсон Д.М. и др. (март 2006 г.). «Использование пиросеквенирования, чтобы пролить свет на микробную экологию глубоких шахт» . БМК Геномика . 7:57 . дои : 10.1186/1471-2164-7-57 . ПМЦ 1483832 . ПМИД 16549033 .
- ^ Томас Т., Гилберт Дж., Мейер Ф. (февраль 2012 г.). «Метагеномика – руководство от выборки к анализу данных» . Микробная информатика и экспериментирование . 2 (1): 3. дои : 10.1186/2042-5783-2-3 . ПМЦ 3351745 . ПМИД 22587947 .
- ^ Бежа О, Сузуки М.Т., Кунин Е.В., Аравинд Л., Хадд А., Нгуен Л.П. и др. (октябрь 2000 г.). «Создание и анализ бактериальных библиотек искусственных хромосом из комплекса морских микробов». Экологическая микробиология . 2 (5): 516–29. Бибкод : 2000EnvMi...2..516B . дои : 10.1046/j.1462-2920.2000.00133.x . ПМИД 11233160 . S2CID 8267748 .
- ^ Jump up to: Перейти обратно: а б Сегата Н., Берниген Д., Тикл Т.Л., Морган XC, Гарретт В.С., Хаттенхауэр С. (май 2013 г.). «Вычислительная метаомика для изучения микробного сообщества» . Молекулярная системная биология . 9 (666): 666. doi : 10.1038/msb.2013.22 . ПМК 4039370 . ПМИД 23670539 .
- ^ Родриг С., Матерна АК, Тимберлейк СК, Блэкберн МК, Мальмстрем РР, Алм Э.Дж., Чисхолм С.В. (июль 2010 г.). Гилберт Дж.А. (ред.). «Открытие секвенирования короткого чтения для метагеномики» . ПЛОС ОДИН . 5 (7): е11840. Бибкод : 2010PLoSO...511840R . дои : 10.1371/journal.pone.0011840 . ПМЦ 2911387 . ПМИД 20676378 .
- ^ Шустер СК (январь 2008 г.). «Секвенирование нового поколения меняет современную биологию». Природные методы . 5 (1): 16–8. дои : 10.1038/nmeth1156 . ПМИД 18165802 . S2CID 1465786 .
- ^ «Метагеномика против закона Мура» . Природные методы . 6 (9): 623. 2009. doi : 10.1038/nmeth0909-623 .
- ^ Стюарт Р.Д., Оффрет М.Д., Уорр А., Уайзер А.Х., Пресс М.О., Лэнгфорд К.В. и др. (февраль 2018 г.). «Сборка 913 микробных геномов в результате метагеномного секвенирования рубца коровы» . Природные коммуникации . 9 (1): 870. Бибкод : 2018NatCo...9..870S . дои : 10.1038/s41467-018-03317-6 . ПМК 5830445 . ПМИД 29491419 .
- ^ Хираока С., Ян CC, Ивасаки В. (сентябрь 2016 г.). «Метагеномика и биоинформатика в микробной экологии: современное состояние и за его пределами» . Микробы и окружающая среда . 31 (3): 204–12. дои : 10.1264/jsme2.ME16024 . ПМК 5017796 . ПМИД 27383682 .
- ^ Перес-Кобас А.Е., Гомес-Валеро Л., Бухризер С. (2020). «Метагеномные подходы в микробной экологии: обновленная информация о полногеномном и маркерном анализе секвенирования генов» . Микробная геномика . 6 (8). дои : 10.1099/mgen.0.000409 . ПМЦ 7641418 . ПМИД 32706331 .
- ^ Jump up to: Перейти обратно: а б Хесс М., Ширба А., Иган Р., Ким Т.В., Чокхавала Х., Шрот Г. и др. (январь 2011 г.). «Метагеномное открытие генов и геномов, разлагающих биомассу, из коровьего рубца». Наука . 331 (6016): 463–7. Бибкод : 2011Sci...331..463H . дои : 10.1126/science.1200387 . ПМИД 21273488 . S2CID 36572885 .
- ^ Цинь Дж., Ли Р., Раес Дж., Арумугам М., Бургдорф К.С., Маничан К. и др. (март 2010 г.). «Каталог генов микробов кишечника человека, созданный с помощью метагеномного секвенирования» . Природа . 464 (7285): 59–65. Бибкод : 2010Natur.464...59. . дои : 10.1038/nature08821 . ПМЦ 3779803 . ПМИД 20203603 . (требуется подписка)
- ^ Jump up to: Перейти обратно: а б Полсон Дж.Н., Стайн О.К., Браво ХК, Поп М. (декабрь 2013 г.). «Дифференциальный анализ численности для исследований микробных маркеров-генов» . Природные методы . 10 (12): 1200–2. дои : 10.1038/nmeth.2658 . ПМК 4010126 . ПМИД 24076764 .
- ^ Jump up to: Перейти обратно: а б с д и ж г Комитет по метагеномике: проблемы и функциональные приложения, Национальный исследовательский совет (2007). Новая наука метагеномика: раскрывая тайны нашей микробной планеты . Вашингтон, округ Колумбия: Издательство национальных академий. дои : 10.17226/11902 . ISBN 978-0-309-10676-4 . ПМИД 21678629 .
- ^ Оулас А., Павлуди С., Полименаку П., Павлопулос Г.А., Папаниколау Н., Котулас Г. и др. (2015). «Метагеномика: инструменты и идеи для анализа данных секвенирования нового поколения, полученных в результате исследований биоразнообразия» . Биоинформатика и биология . 9 : 75–88. дои : 10.4137/BBI.S12462 . ПМЦ 4426941 . ПМИД 25983555 .
- ^ Менде Д.Р., Уоллер А.С., Сунагава С., Ярвелин А.И., Чан М.М., Арумугам М. и др. (23 февраля 2012 г.). «Оценка метагеномной сборки с использованием смоделированных данных секвенирования следующего поколения» . ПЛОС ОДИН . 7 (2): e31386. Бибкод : 2012PLoSO...731386M . дои : 10.1371/journal.pone.0031386 . ПМЦ 3285633 . ПМИД 22384016 .
- ^ Бальцер С., Мальде К., Громе М.А., Йонассен И. (апрель 2013 г.). «Фильтрация повторяющихся чтений из 454 данных пиросеквенирования» . Биоинформатика . 29 (7): 830–6. doi : 10.1093/биоинформатика/btt047 . ПМЦ 3605598 . ПМИД 23376350 .
- ^ Мохаммед М.Х., Чадарам С., Командури Д., Гош Т.С., Манде С.С. (сентябрь 2011 г.). «Eu-Detect: алгоритм обнаружения эукариотических последовательностей в наборах метагеномных данных». Журнал биологических наук . 36 (4): 709–17. дои : 10.1007/s12038-011-9105-2 . ПМИД 21857117 . S2CID 25857874 .
- ^ Шмидер Р., Эдвардс Р. (март 2011 г.). «Быстрая идентификация и удаление загрязнений последовательностей из наборов геномных и метагеномных данных» . ПЛОС ОДИН . 6 (3): e17288. Бибкод : 2011PLoSO...617288S . дои : 10.1371/journal.pone.0017288 . ПМК 3052304 . ПМИД 21408061 .
- ^ Jump up to: Перейти обратно: а б с д и Кунин В., Коупленд А., Лапидус А., Мавроматис К., Хугенхольц П. (декабрь 2008 г.). «Руководство биоинформатика по метагеномике» . Обзоры микробиологии и молекулярной биологии . 72 (4): 557–78, Оглавление. дои : 10.1128/MMBR.00009-08 . ПМЦ 2593568 . ПМИД 19052320 .
- ^ Намики Т., Хачия Т., Танака Х., Сакакибара Ю. (ноябрь 2012 г.). «MetaVelvet: расширение ассемблера Velvet для сборки метагенома de novo из чтения коротких последовательностей» . Исследования нуклеиновых кислот . 40 (20): е155. дои : 10.1093/nar/gks678 . ПМК 3488206 . ПМИД 22821567 .
- ^ Зербино Д.Р., Бирни Э. (май 2008 г.). «Бархат: алгоритмы сборки короткого чтения de novo с использованием графов де Брёйна» . Геномные исследования . 18 (5): 821–9. дои : 10.1101/гр.074492.107 . ПМК 2336801 . ПМИД 18349386 .
- ^ Бертон Дж. Н., Лячко И., Данэм М. Дж., Шендюр Дж. (май 2014 г.). «Деконволюция сборок метагенома на уровне вида с помощью карт вероятности контакта на основе Hi-C» . Г3 . 4 (7): 1339–46. дои : 10.1534/g3.114.011825 . ПМЦ 4455782 . ПМИД 24855317 .
- ^ Jump up to: Перейти обратно: а б Хьюсон Д.Х., Митра С., Рушевей Х.Дж., Вебер Н., Шустер С.К. (сентябрь 2011 г.). «Интегративный анализ последовательностей окружающей среды с использованием MEGAN4» . Геномные исследования . 21 (9): 1552–60. дои : 10.1101/гр.120618.111 . ПМК 3166839 . ПМИД 21690186 .
- ^ Чжу В., Ломсадзе А., Бородовский М. (июль 2010 г.). «Идентификация генов Ab initio в метагеномных последовательностях» . Исследования нуклеиновых кислот . 38 (12): е132. дои : 10.1093/nar/gkq275 . ПМЦ 2896542 . ПМИД 20403810 .
- ^ Конопка А (ноябрь 2009 г.). «Что такое экология микробного сообщества?» . Журнал ISME . 3 (11): 1223–30. Бибкод : 2009ISMEJ...3.1223K . дои : 10.1038/ismej.2009.88 . ПМИД 19657372 .
- ^ Jump up to: Перейти обратно: а б Хьюсон Д.Х., Ош А.Ф., Ци Дж, Шустер СК (март 2007 г.). «МЕГАН-анализ метагеномных данных» . Геномные исследования . 17 (3): 377–86. дои : 10.1101/гр.5969107 . ПМЦ 1800929 . ПМИД 17255551 .
- ^ Сегата Н., Уолдрон Л., Балларини А., Нарасимхан В., Юссон О., Хаттенхауэр К. (июнь 2012 г.). «Метагеномное профилирование микробного сообщества с использованием уникальных маркерных генов, специфичных для клады» . Природные методы . 9 (8): 811–4. дои : 10.1038/nmeth.2066 . ПМЦ 3443552 . ПМИД 22688413 .
- ^ Сунагава С., Менде Д.Р., Целлер Г., Искьердо-Карраско Ф., Бергер С.А., Култима Дж.Р. и др. (декабрь 2013 г.). «Метагеномное профилирование видов с использованием универсальных филогенетических маркерных генов». Природные методы . 10 (12): 1196–9. дои : 10.1038/nmeth.2693 . ПМИД 24141494 . S2CID 7728395 .
- ^ Jump up to: Перейти обратно: а б Миланезе А., Менде Д.Р., Паоли Л., Салазар Г., Рушевей Х.Дж., Куэнка М. и др. (март 2019 г.). «Микробная численность, активность и геномное профилирование популяции с помощью mOTUs2» . Природные коммуникации . 10 (1): 1014. Бибкод : 2019NatCo..10.1014M . дои : 10.1038/s41467-019-08844-4 . ПМК 6399450 . ПМИД 30833550 .
- ^ Лю Б., Гиббонс Т., Годси М., Треанген Т., Поп М. (2011). «Точная и быстрая оценка таксономических профилей на основе метагеномных последовательностей» . БМК Геномика . 12 (Приложение 2): S4. дои : 10.1186/1471-2164-12-S2-S4 . ПМК 3194235 . ПМИД 21989143 .
- ^ Дади Т.Х., Ренард Б.Ю., Вилер Л.Х., Земмлер Т., Райнерт К. (2017). «SLIMM: видовая идентификация микроорганизмов по метагеномам» . ПерДж . 5 : е3138. дои : 10.7717/peerj.3138 . ПМЦ 5372838 . ПМИД 28367376 .
- ^ Пагани И., Лиолиос К., Янссон Дж., Чен И.М., Смирнова Т., Носрат Б. и др. (январь 2012 г.). «Онлайн-база данных Genomes (GOLD) v.4: состояние геномных и метагеномных проектов и связанные с ними метаданные» . Исследования нуклеиновых кислот . 40 (Проблема с базой данных): D571-9. дои : 10.1093/nar/gkr1100 . ПМК 3245063 . ПМИД 22135293 .
- ^ Мейер Ф., Паарманн Д., Д'Суза М., Олсон Р., Гласс Э.М., Кубал М. и др. (сентябрь 2008 г.). «RAST-сервер метагеномики — общедоступный ресурс для автоматического филогенетического и функционального анализа метагеномов» . БМК Биоинформатика . 9 : 386. дои : 10.1186/1471-2105-9-386 . ПМК 2563014 . ПМИД 18803844 .
- ^ Марковиц В.М., Чен И.М., Чу К., Сето Е., Паланиаппан К., Гречкин Ю. и др. (январь 2012 г.). «IMG/M: интегрированная система управления метагеномными данными и сравнительного анализа» . Исследования нуклеиновых кислот . 40 (Проблема с базой данных): D123-9. дои : 10.1093/nar/gkr975 . ПМК 3245048 . ПМИД 22086953 .
- ^ Jump up to: Перейти обратно: а б Митра С., Рупек П., Рихтер Д.С., Урих Т., Гилберт Дж.А., Мейер Ф. и др. (февраль 2011 г.). «Функциональный анализ метагеномов и метатранскриптомов с использованием SEED и KEGG» . БМК Биоинформатика . 12 (Приложение 1): S21. дои : 10.1186/1471-2105-12-S1-S21 . ПМК 3044276 . ПМИД 21342551 .
- ^ Бенсон Д.А., Кавано М., Кларк К., Карш-Мизрачи И., Липман Дж., Остелл Дж., Сэйерс Э.В. (январь 2013 г.). «ГенБанк» . Исследования нуклеиновых кислот . 41 (Проблема с базой данных): D36-42. дои : 10.1093/nar/gks1195 . ПМК 3531190 . ПМИД 23193287 .
- ^ Базинет А.Л., член парламента Каммингса (май 2012 г.). «Сравнительная оценка программ классификации последовательностей» . БМК Биоинформатика . 13:92 . дои : 10.1186/1471-2105-13-92 . ПМЦ 3428669 . ПМИД 22574964 .
- ^ Оунит Р., Ванамакер С., Клоуз Т.Дж., Лонарди С. (март 2015 г.). «CLARK: быстрая и точная классификация метагеномных и геномных последовательностей с использованием дискриминационных k-меров» . БМК Геномика . 16 (1): 236. дои : 10.1186/s12864-015-1419-2 . ПМЦ 4428112 . ПМИД 25879410 .
- ^ Пратас Д., Пиньо А.Дж., Силва Р.М., Родригес Х.М., Хоссейни М., Каэтано Т., Феррейра П.Дж. (февраль 2018 г.). «СОКОЛ: метод определения метагеномного состава древней ДНК». bioRxiv 10.1101/267179 .
- ^ Курокава К., Ито Т., Кувахара Т., Осима К., Тох Х., Тойода А. и др. (август 2007 г.). «Сравнительная метагеномика выявила обычно обогащенные наборы генов в микробиомах кишечника человека» . Исследование ДНК . 14 (4): 169–81. дои : 10.1093/dnares/dsm018 . ПМЦ 2533590 . ПМИД 17916580 .
- ^ Jump up to: Перейти обратно: а б с д и ж Саймон С., Дэниел Р. (февраль 2011 г.). «Метагеномный анализ: тенденции прошлого и будущего» . Прикладная и экологическая микробиология . 77 (4): 1153–61. Бибкод : 2011ApEnM..77.1153S . дои : 10.1128/АЕМ.02345-10 . ПМК 3067235 . ПМИД 21169428 .
- ^ Уиллнер Д., Тербер Р.В., Ровер Ф. (июль 2009 г.). «Метагеномные признаки 86 микробных и вирусных метагеномов» . Экологическая микробиология . 11 (7): 1752–66. Бибкод : 2009EnvMi..11.1752W . дои : 10.1111/j.1462-2920.2009.01901.x . ПМИД 19302541 .
- ^ Гош Т.С., Мохаммед М.Х., Раджасингх Х., Чадарам С., Манде С.С. (2011). «HabiSign: новый подход к сравнению метагеномов и быстрой идентификации последовательностей, специфичных для среды обитания» . БМК Биоинформатика . 12 Приложение 13 (Дополнение 13): С9. дои : 10.1186/1471-2105-12-s13-s9 . ПМЦ 3278849 . ПМИД 22373355 .
- ^ Фимерели Д., Объезды В., Конопка Т. (апрель 2013 г.). «TriageTools: инструменты для разделения и определения приоритетов анализа данных высокопроизводительного секвенирования» . Исследования нуклеиновых кислот . 41 (7): е86. дои : 10.1093/нар/gkt094 . ПМЦ 3627586 . ПМИД 23408855 .
- ^ Майе Н., Леметр С., Чихи Р., Лавенье Д., Петерлонго П. (2012). «Сравнивает: сравнение огромных метагеномных экспериментов» . БМК Биоинформатика . 13 (Приложение 19): S10. дои : 10.1186/1471-2105-13-S19-S10 . ПМЦ 3526429 . ПМИД 23282463 .
- ^ Кунтал Б.К., Гош Т.С., Манде С.С. (октябрь 2013 г.). «Сообщество-анализатор: платформа для визуализации и сравнения структуры микробного сообщества в разных микробиомах» . Геномика . 102 (4): 409–18. дои : 10.1016/j.ygeno.2013.08.004 . ПМИД 23978768 .
- ^ Вернер Дж.Дж., Найтс Д., Гарсия М.Л., Скалфоне Н.Б., Смит С., Ярашески К. и др. (март 2011 г.). «Структуры бактериального сообщества уникальны и устойчивы в полномасштабных биоэнергетических системах» . Труды Национальной академии наук Соединенных Штатов Америки . 108 (10): 4158–63. Бибкод : 2011PNAS..108.4158W . дои : 10.1073/pnas.1015676108 . ПМК 3053989 . ПМИД 21368115 .
- ^ Макинерни MJ, Зибер-младший, Gunsalus RP (декабрь 2009 г.). «Синтрофия в анаэробных глобальных углеродных циклах» . Современное мнение в области биотехнологии . 20 (6): 623–32. дои : 10.1016/j.copbio.2009.10.001 . ПМК 2790021 . ПМИД 19897353 .
- ^ Клитгорд Н., Сегре Д. (август 2011 г.). «Экосистемная биология микробного метаболизма». Современное мнение в области биотехнологии . 22 (4): 541–6. дои : 10.1016/j.copbio.2011.04.018 . ПМИД 21592777 .
- ^ Лейнингер С., Урих Т., Шлотер М., Шварк Л., Ци Дж., Никол Г.В. и др. (август 2006 г.). «Среди прокариот, окисляющих аммиак, в почвах преобладают археи». Природа . 442 (7104): 806–9. Бибкод : 2006Natur.442..806L . дои : 10.1038/nature04983 . ПМИД 16915287 . S2CID 4380804 .
- ^ Паес-Эспино Д., Элое-Фадрош Е.А., Павлопулос Г.А., Томас А.Д., Хантеманн М., Михайлова Н. и др. (август 2016 г.). «Открытие земного вирома » Природа 536 (7617): 425–30. Бибкод : 2016Nature.536..425P . дои : 10.1038/nature19094 . ПМИД 27533034 . S2CID 4466854 .
- ^ Паес-Эспино Д., Чен И.А., Паланиаппан К., Ратнер А., Чу К., Сето Е. и др. (январь 2017 г.). «IMG/VR: база данных культивируемых и некультивируемых ДНК-вирусов и ретровирусов» . Исследования нуклеиновых кислот . 45 (Д1): Д457–Д465. дои : 10.1093/nar/gkw1030 . ПМК 5210529 . ПМИД 27799466 .
- ^ Паес-Эспино Д., Ру С., Чен И.А., Паланиаппан К., Ратнер А., Чу К. и др. (январь 2019 г.). «IMG/VR v.2.0: интегрированная система управления и анализа данных для культивируемых и находящихся в окружающей среде вирусных геномов» . Исследования нуклеиновых кислот . 47 (Д1): Д678–Д686. дои : 10.1093/nar/gky1127 . ПМК 6323928 . ПМИД 30407573 .
- ^ Паес-Эспино Д., Павлопулос Г.А., Иванова Н.Н., Кирпидес Н.К. (август 2017 г.). «Конвейер обнаружения нецелевых последовательностей вирусов и кластеризация вирусов для метагеномных данных» (PDF) . Протоколы природы . 12 (8): 1673–1682. дои : 10.1038/nprot.2017.063 . ПМИД 28749930 . S2CID 2127494 .
- ^ Кристенсен Д.М., Мушегян А.Р., Доля В.В., Кунин Е.В. (январь 2010 г.). «Новые измерения мира вирусов, открытые с помощью метагеномики» . Тенденции в микробиологии . 18 (1): 11–9. дои : 10.1016/j.tim.2009.11.003 . ПМЦ 3293453 . ПМИД 19942437 .
- ^ Керепеси С, Гролмуш В (март 2016 г.). «Гигантские вирусы пустыни Кач». Архив вирусологии . 161 (3): 721–4. arXiv : 1410.1278 . дои : 10.1007/s00705-015-2720-8 . ПМИД 26666442 . S2CID 13145926 .
- ^ Керепеси С, Гролмуш В (июнь 2017 г.). «Система поиска гигантских вирусов обнаружила множество гигантских вирусов в сухих долинах Антарктики». Архив вирусологии . 162 (6): 1671–1676. arXiv : 1503.05575 . дои : 10.1007/s00705-017-3286-4 . ПМИД 28247094 . S2CID 1925728 .
- ^ Copeland CS (сентябрь – октябрь 2017 г.). «Мир внутри нас» (PDF) . Журнал здравоохранения Нового Орлеана : 21–26.
- ^ Янссон Дж (2011). «На пути к «Тера-Терра»: терабазное секвенирование земных метагеномов. Печать по электронной почте» . Микроб . Том. 6, нет. 7. с. 309. Архивировано из оригинала 31 марта 2012 года.
- ^ Фогель Т.М., Симонет П., Янссон Дж.К., Хирш П.Р., Тидже Дж.М., Ван Эльсас Дж.Д., Бэйли М.Дж., Налин Р., Филиппот Л. (2009). «TerraGenome: Консорциум по секвенированию метагенома почвы» . Обзоры природы Микробиология . 7 (4): 252. doi : 10.1038/nrmicro2119 .
- ^ «Домашняя страница TerraGenome» . Международный консорциум секвенирования TerraGenome . Проверено 30 декабря 2011 г.
- ^ Jump up to: Перейти обратно: а б Комитет по метагеномике: проблемы и функциональные приложения, Национальный исследовательский совет (2007). Понимание нашей микробной планеты: новая наука метагеномика (PDF) . Пресса национальных академий. Архивировано из оригинала (PDF) 30 октября 2012 года . Проверено 30 декабря 2011 г.
- ^ Чарльз Т. (2010). «Возможности исследования растительно-микробных взаимодействий методами метагеномики». Метагеномика: теория, методы и приложения . Кайстер Академик Пресс. ISBN 978-1-904455-54-7 .
- ^ Брингель Ф., Куэ I (22 мая 2015 г.). «Основная роль микроорганизмов филлосферы на стыке между функционированием растений и динамикой газовых примесей в атмосфере» . Границы микробиологии . 6 : 486. дои : 10.3389/fmicb.2015.00486 . ПМК 4440916 . ПМИД 26052316 .
- ^ Ли ЛЛ, Маккоркл С.Р., Мончи С., Тагави С., ван дер Лели Д. (май 2009 г.). «Биоразведка метагеномов: гликозилгидролазы для преобразования биомассы» . Биотехнология для биотоплива . 2:10 . дои : 10.1186/1754-6834-2-10 . ПМК 2694162 . ПМИД 19450243 .
- ^ Йенике С., Андер С., Бекель Т., Бисдорф Р., Дрёге М., Гартеманн К.Х. и др. (январь 2011 г.). Азиз РК (ред.). «Сравнительный и совместный анализ двух наборов метагеномных данных из биогазового ферментера, полученных с помощью 454-пиросеквенирования» . ПЛОС ОДИН . 6 (1): e14519. Бибкод : 2011PLoSO...614519J . дои : 10.1371/journal.pone.0014519 . ПМК 3027613 . ПМИД 21297863 .
- ^ Суен Дж., Скотт Дж.Дж., Эйлуорд Ф.О., Адамс С.М., Тринге С.Г., Пинто-Томас А.А. и др. (сентябрь 2010 г.). Зонненбург Дж. (ред.). «Микробиом насекомых-травоядных с высокой способностью к разложению биомассы растений» . ПЛОС Генетика . 6 (9): e1001129. дои : 10.1371/journal.pgen.1001129 . ПМЦ 2944797 . ПМИД 20885794 .
- ^ Саймон С., Дэниел Р. (ноябрь 2009 г.). «Достижения и новые знания, раскрытые метагеномными подходами» . Прикладная микробиология и биотехнология . 85 (2): 265–76. дои : 10.1007/s00253-009-2233-z . ПМЦ 2773367 . ПМИД 19760178 .
- ^ Вонг Д. (2010). «Применение метагеномики для промышленных биопродуктов». Метагеномика: теория, методы и приложения . Кайстер Академик Пресс. ISBN 978-1-904455-54-7 .
- ^ Jump up to: Перейти обратно: а б Шлосс П.Д., Хандельсман Дж. (июнь 2003 г.). «Биотехнологические перспективы метагеномики» (PDF) . Современное мнение в области биотехнологии . 14 (3): 303–10. дои : 10.1016/S0958-1669(03)00067-3 . ПМИД 12849784 . Архивировано из оригинала (PDF) 4 марта 2016 года . Проверено 20 января 2012 г.
- ^ Jump up to: Перейти обратно: а б с Какирде К.С., Петрушка LC, Лайлз М.Р. (ноябрь 2010 г.). «Размер имеет значение: прикладные подходы к метагеномике почвы» . Биология и биохимия почвы . 42 (11): 1911–1923. doi : 10.1016/j.soilbio.2010.07.021 . ПМЦ 2976544 . ПМИД 21076656 .
- ^ Парачин Н.С., Горва-Грауслунд М.Ф. (май 2011 г.). «Выделение ксилозоизомераз путем скрининга на основе последовательностей и функций из метагеномной библиотеки почвы» . Биотехнология для биотоплива . 4 (1): 9. дои : 10.1186/1754-6834-4-9 . ПМК 3113934 . ПМИД 21545702 .
- ^ Ховер Б.М., Ким Ш., Кац М., Чарлоп-Пауэрс З., Оуэн Дж.Г., Терней М.А. и др. (апрель 2018 г.). «Независимое от культуры открытие малацидинов как кальций-зависимых антибиотиков, обладающих активностью против грамположительных патогенов с множественной лекарственной устойчивостью» . Природная микробиология . 3 (4): 415–422. дои : 10.1038/s41564-018-0110-1 . ПМК 5874163 . ПМИД 29434326 .
- ^ Раес Дж., Летунич И., Ямада Т., Дженсен Л.Дж., Борк П. (март 2011 г.). «На пути к экологии, основанной на молекулярных признаках, посредством интеграции биогеохимических, географических и метагеномных данных» . Молекулярная системная биология . 7 : 473. дои : 10.1038/msb.2011.6 . ПМК 3094067 . ПМИД 21407210 .
- ^ Лавери Т.Дж., Роуднью Б., Сеймур Дж., Митчелл Дж.Г., Джеффрис Т. (2012). Стейнке Д. (ред.). «В микробном метагеноме фекалий австралийского морского льва (Neophoca cinerea) обнаружен высокий потенциал транспортировки и круговорота питательных веществ» . ПЛОС ОДИН . 7 (5): e36478. Бибкод : 2012PLoSO...736478L . дои : 10.1371/journal.pone.0036478 . ПМК 3350522 . ПМИД 22606263 .
- ^ «Что плавает в реке? Просто ищите ДНК» . NPR.org . 24 июля 2013 года . Проверено 10 октября 2014 г.
- ^ Чуа, Физилия Ю.С.; Крэмптон-Платт, Алекс; Ламмерс, Юрий; Алсос, Ингер Г.; Боссенкул, Санне; Боманн, Кристина (25 мая 2021 г.). «Метагеномика: жизнеспособный инструмент для реконструкции рациона травоядных» . Ресурсы молекулярной экологии . 21 (7): 1755–0998.13425. дои : 10.1111/1755-0998.13425 . ПМЦ 8518049 . ПМИД 33971086 .
- ^ Чуа, Физилия Ю.С.; Карё, Кристиан; Крэмптон-Платт, Алекс; Рейес-Авила, Клаудия С.; Джонс, Гарет; Штрайкер, Дэниел Г.; Боманн, Кристина (4 июля 2022 г.). «Двухэтапный метагеномный подход к идентификации и сборке контигов митохондриальной ДНК позвоночных жертв из крови обыкновенных летучих мышей-вампиров (Desmodus rotundus)» . Метабаркодирование и метагеномика . 6 : e78756. дои : 10.3897/mbmg.6.78756 . ISSN 2534-9708 . S2CID 248041252 .
- ^ Георг I, Стенуит Б, Агатос С.Н. (2010). «Применение метагеномики к биоремедиации». В Марко Д. (ред.). Метагеномика: теория, методы и приложения . Кайстер Академик Пресс. ISBN 978-1-904455-54-7 .
- ^ Циммер С (13 июля 2010 г.). «Как микробы защищают и определяют нас» . Нью-Йорк Таймс . Проверено 29 декабря 2011 г.
- ^ Нельсон К.Е. и Уайт Б.А. (2010). «Метагеномика и ее приложения к изучению микробиома человека». Метагеномика: теория, методы и приложения . Кайстер Академик Пресс. ISBN 978-1-904455-54-7 .
- ^ Jump up to: Перейти обратно: а б Цинь, Цзюньцзе; Ли, Жуйцян; Раес, Йерун; Арумугам, Маниможиян; Бургдорф, Кристоффер Солвстен; Маничан, Чайсаван; Нильсен, Трина; Понс, Николас; Левенес, Флоренция; Ямада, Такудзи; Менде, Дэниел Р.; Ли, Цзюньхуа; Сюй, Цзюньмин; Ли, Шаочуань; Ли, Дунфан (2010). «Каталог генов микробов кишечника человека, созданный с помощью метагеномного секвенирования» . Природа . 464 (7285): 59–65. Бибкод : 2010Natur.464...59. . дои : 10.1038/nature08821 . ISSN 1476-4687 . ПМЦ 3779803 . ПМИД 20203603 .
- ^ Цинь Дж., Ли Р., Раес Дж., Арумугам М., Бургдорф К.С., Маничан К. и др. (март 2010 г.). «Каталог генов микробов кишечника человека, созданный с помощью метагеномного секвенирования» . Природа . 464 (7285): 59–65. Бибкод : 2010Natur.464...59. . дои : 10.1038/nature08821 . ПМЦ 3779803 . ПМИД 20203603 .
- ^ Абубакер, Сахар; Сегата, Никола; Голл, Йоханнес; Шуберт, Александрия М.; Изард, Жак; Кантарел, Брэнди Л.; Родригес-Мюллер, Бельтран; Цукер, Джереми; Тиагараджан, Матанги; Анрисса, Бернар; Уайт, Оуэн; Келли, Скотт Т.; Мете, Барбара; Шлосс, Патрик Д.; Геверс, Дирк; Митрева, Македонка; Хаттенхауэр, Кертис (2012). «Вычислительная биология PLOS: метаболическая реконструкция метагеномных данных и ее применение к микробиому человека» . PLOS Вычислительная биология . 8 (6): e1002358. Бибкод : 2012PLSCB...8E2358A . дои : 10.1371/journal.pcbi.1002358 . ПМЦ 3374609 . ПМИД 22719234 .
- ^ Чуа, Физилия Ин Ши; Расмуссен, Джейкоб Агербо (11 мая 2022 г.). «Возьмем метагеномику под свое крыло» . Обзоры природы Микробиология . 20 (8): 447. doi : 10.1038/s41579-022-00746-5 . ISSN 1740-1534 . ПМИД 35546350 . S2CID 248739527 .
- ^ Чиу, Чарльз Ю.; Миллер, Стивен А. (2019). «Клиническая метагеномика» . Обзоры природы Генетика . 20 (6): 341–355. дои : 10.1038/s41576-019-0113-7 . ISSN 1471-0064 . ПМЦ 6858796 . ПМИД 30918369 .
- ^ Закшевский М., Рашич Г., Дарбро Дж., Краузе Л., Пу Ю.С., Филипович И. и др. (2018). «Картирование вирома у выловленных в дикой природе Aedes aegypti из Кэрнса и Бангкока» . Научный представитель . 8 (1): 4690. Бибкод : 2018NatSR...8.4690Z . дои : 10.1038/s41598-018-22945-y . ПМК 5856816 . ПМИД 29549363 .
- ^ Тендель М (2020). «Целевая метагеномика дает представление о потенциальных клещевых патогенах» . J Clin Микробиол . 58 (11). дои : 10.1128/JCM.01893-20 . ПМЦ 7587107 . ПМИД 32878948 .
- ^ Парри Р., Джеймс М.Э., Асгари С. (2021). «Раскрытие всемирного разнообразия и эволюции вирома комаров Aedes aegypti и Aedes albopictus» . Микроорганизмы . 9 (8): 1653. doi : 10.3390/microorganisms9081653 . ПМЦ 8398489 . ПМИД 34442732 .
- ^ Батовска Дж., Ми П.Т., Линч С.Е., Собридж Т.И., Родони BC (2019). «Чувствительность и специфичность метатранскриптомики как инструмента наблюдения за арбовирусами» . Научный представитель . 9 (1): 19398. Бибкод : 2019НатСР...919398Б . дои : 10.1038/s41598-019-55741-3 . ПМК 6920425 . ПМИД 31852942 .
- ^ Батовска Дж., Линч С.Э., Родони Б.К., Собридж Т.И., Коган Н.О. (2017). «Метагеномное обнаружение арбовирусов с использованием секвенирования нанопор MinION» . Дж. Вироловые методы . 249 : 79–84. дои : 10.1016/j.jviromet.2017.08.019 . ПМИД 28855093 .
Внешние ссылки [ править ]
- В центре внимания метагеномика на Nature Reviews Microbiology сайте журнала
- Инициатива «Критическая оценка интерпретации метагенома» (CAMI) по оценке методов метагеномики.