Биоинформатика


Биоинформатика ( / ˌ b aɪ . oʊ ˌ ɪ n f ər ˈ m æ t ɪ k s / ) — междисциплинарная область науки , которая разрабатывает методы и программные средства для понимания биологических данных, особенно когда наборы данных большие и сложные. Биоинформатика использует биологию , химию , физику , информатику , компьютерное программирование , информационную инженерию , математику и статистику для анализа и интерпретации биологических данных . [1] Последующий процесс анализа и интерпретации данных называется вычислительной биологией .
Вычислительные, статистические методы и методы компьютерного программирования использовались для компьютерного моделирования биологических запросов. Они включают повторно используемые специфические «конвейеры» анализа, особенно в области геномики , например, путем идентификации генов и однонуклеотидных полиморфизмов ( SNP ). Эти конвейеры используются для лучшего понимания генетической основы болезней, уникальных адаптаций, желаемых свойств (особенно у сельскохозяйственных видов) или различий между популяциями. Биоинформатика также включает протеомику , которая пытается понять принципы организации последовательностей нуклеиновых кислот и белков . [2]
Обработка изображений и сигналов позволяет извлекать полезные результаты из больших объемов необработанных данных. В области генетики он помогает секвенировать и аннотировать геномы и их наблюдаемые мутации . Биоинформатика включает в себя анализ текста биологической литературы и разработку биологических и генных онтологий для организации и запроса биологических данных. Он также играет роль в анализе экспрессии и регуляции генов и белков. Инструменты биоинформатики помогают сравнивать, анализировать и интерпретировать генетические и геномные данные и, в более общем плане, понимать эволюционные аспекты молекулярной биологии. На более интегративном уровне это помогает анализировать и каталогизировать биологические пути и сети, которые являются важной частью системной биологии . В структурной биологии он помогает в моделировании ДНК. [3] РНК, [3] [4] белки [5] а также биомолекулярные взаимодействия. [6] [7] [8] [9]
История [ править ]
Первое определение термина «биоинформатика» было предложено Паулином Хогевегом и Беном Хеспером в 1970 году для обозначения изучения информационных процессов в биотических системах. [10] [11] [12] [13] [14] Это определение помещало биоинформатику в область, параллельную биохимии (изучение химических процессов в биологических системах). [11]
Биоинформатика и вычислительная биология включали анализ биологических данных, особенно последовательностей ДНК, РНК и белков. Область биоинформатики пережила взрывной рост, начиная с середины 1990-х годов, во многом благодаря проекту «Геном человека» и быстрому развитию технологии секвенирования ДНК. [ нужна ссылка ]
Анализ биологических данных для получения значимой информации включает в себя написание и запуск программ, использующих алгоритмы теории графов , искусственного интеллекта , мягких вычислений , интеллектуального анализа данных , обработки изображений и компьютерного моделирования . Алгоритмы, в свою очередь, зависят от теоретических основ, таких как дискретная математика , теория управления , теория систем , теория информации и статистика . [ нужна ссылка ]
Последовательности [ править ]


С момента завершения проекта «Геном человека» произошел огромный прогресс в скорости и сокращении затрат: некоторые лаборатории смогли секвенировать более 100 000 миллиардов оснований каждый год, а полный геном можно секвенировать за 1000 долларов или меньше. [15]
Компьютеры стали играть важную роль в молекулярной биологии, когда последовательности белков стали доступны после того, как Фредерик Сэнгер определил последовательность инсулина в начале 1950-х годов. [16] [17] Сравнение нескольких последовательностей вручную оказалось непрактичным. Маргарет Окли Дэйхофф , пионер в этой области, [18] составил одну из первых баз данных последовательностей белков, первоначально опубликованную в виде книг. [19] а также методы выравнивания последовательностей и молекулярной эволюции . [20] Еще одним ранним вкладчиком в биоинформатику был Элвин А. Кабат , который впервые ввел анализ биологических последовательностей в 1970 году, опубликовав обширные тома последовательностей антител вместе с Тай Тэ Ву в период с 1980 по 1991 год. [21]
В 1970-х годах к бактериофагам MS2 и øX174 были применены новые методы секвенирования ДНК, а расширенные нуклеотидные последовательности затем были проанализированы с помощью информационных и статистических алгоритмов. Эти исследования показали, что хорошо известные особенности, такие как сегменты кодирования и триплетный код, выявляются при простом статистическом анализе и стали доказательством концепции того, что биоинформатика будет проницательной. [22] [23]
Цели [ править ]
Чтобы изучить, как нормальная клеточная активность изменяется при различных болезненных состояниях, необходимо объединить необработанные биологические данные, чтобы сформировать полную картину этой активности. Поэтому [ когда? ] , область биоинформатики развилась таким образом, что наиболее насущная задача теперь связана с анализом и интерпретацией различных типов данных. Сюда также входят нуклеотидные и аминокислотные последовательности , белковые домены и белковые структуры . [24]
Важные субдисциплины биоинформатики и вычислительной биологии включают:
- Разработка и внедрение компьютерных программ для эффективного доступа, управления и использования различных типов информации.
- Разработка новых математических алгоритмов и статистических мер для оценки взаимосвязей между членами больших наборов данных. Например, существуют способы локализации гена внутри последовательности, прогнозирования структуры и/или функции белка и группировки белковых последовательностей в семейства родственных последовательностей.
Основная цель биоинформатики — улучшить понимание биологических процессов. Что отличает его от других подходов, так это его ориентация на разработку и применение вычислительно-интенсивных методов для достижения этой цели. Примеры включают: распознавание образов , интеллектуальный анализ данных , алгоритмы машинного обучения и визуализацию . Основные исследовательские усилия в этой области включают выравнивание последовательностей , поиск генов , сборку генома , разработку лекарств , открытие лекарств , выравнивание структуры белков , предсказание структуры белков , предсказание экспрессии генов и белок-белковых взаимодействий , полногеномные исследования ассоциаций , моделирование эволюции. и деление клеток/митоз.
Биоинформатика влечет за собой создание и развитие баз данных, алгоритмов, вычислительных и статистических методов, а также теории для решения формальных и практических проблем, возникающих в результате управления и анализа биологических данных.
За последние несколько десятилетий быстрое развитие геномных и других технологий молекулярных исследований, а также развитие информационных технологий в совокупности привело к появлению огромного количества информации, связанной с молекулярной биологией. Биоинформатика — это название, данное математическим и вычислительным подходам, используемым для понимания биологических процессов.
Обычные действия в биоинформатике включают картирование и анализ последовательностей ДНК и белков, выравнивание последовательностей ДНК и белков для их сравнения, а также создание и просмотр трехмерных моделей белковых структур.
Анализ последовательности [ править ]
Поскольку бактериофаг Phage Φ-X174 был секвенирован в 1977 г. [25] последовательности ДНК тысяч организмов были расшифрованы и сохранены в базах данных. Эта информация о последовательностях анализируется для определения генов, которые кодируют белки , гены РНК, регуляторные последовательности, структурные мотивы и повторяющиеся последовательности. Сравнение генов внутри одного вида или между разными видами может показать сходство функций белков или отношения между видами (использование молекулярной систематики для построения филогенетических деревьев ). С ростом объема данных анализ последовательностей ДНК вручную уже давно стал непрактичным. Компьютерные программы , такие как BLAST, регулярно используются для поиска последовательностей — по состоянию на 2008 год из более чем 260 000 организмов, содержащих более 190 миллиардов нуклеотидов . [26]
Секвенирование ДНК [ править ]
Прежде чем последовательности можно будет проанализировать, их получают из банка хранения данных, такого как GenBank. Секвенирование ДНК по-прежнему остается нетривиальной проблемой, поскольку необработанные данные могут быть зашумлены или на них могут влиять слабые сигналы. алгоритмы Были разработаны определения оснований для различных экспериментальных подходов к секвенированию ДНК.
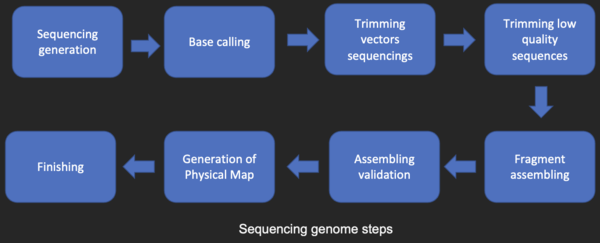
Последовательность сборки [ править ]
Большинство методов секвенирования ДНК позволяют получить короткие фрагменты последовательности, которые необходимо собрать для получения полных последовательностей гена или генома. Техника секвенирования дробовиком (используется Институтом геномных исследований (TIGR) для секвенирования первого бактериального генома Haemophilus influenzae ) [27] генерирует последовательности многих тысяч небольших фрагментов ДНК (длиной от 35 до 900 нуклеотидов, в зависимости от технологии секвенирования). Концы этих фрагментов перекрываются, и при правильном выравнивании с помощью программы сборки генома их можно использовать для реконструкции полного генома. Секвенирование методом дробовика позволяет быстро получить данные о последовательностях, но задача сборки фрагментов может быть довольно сложной для более крупных геномов. Для генома такого размера, как геном человека , на многопроцессорных компьютерах с большой памятью может потребоваться много дней процессорного времени, чтобы собрать фрагменты, и полученная сборка обычно содержит множество пробелов, которые необходимо заполнить позже. Секвенирование дробовика — метод выбора практически для всех секвенированных геномов (а не методы обрыва цепи или химической деградации), а алгоритмы сборки генома являются важной областью исследований в области биоинформатики.
Аннотация генома [ править ]
В геномике аннотация . относится к процессу маркировки стоп- и стартовых областей генов и других биологических особенностей в секвенированной последовательности ДНК Многие геномы слишком велики, чтобы их можно было аннотировать вручную. Поскольку скорость секвенирования превышает скорость аннотации генома, аннотация генома стала новым узким местом в биоинформатике. [ когда? ] .
Аннотацию генома можно разделить на три уровня: уровень нуклеотидов , белков и уровень процессов.
Обнаружение генов является главным аспектом аннотации на уровне нуклеотидов. Для сложных геномов может оказаться успешным сочетание прогнозирования генов ab initio и сравнения последовательностей с базами данных экспрессируемых последовательностей и другими организмами. Аннотация на уровне нуклеотидов также позволяет интегрировать последовательность генома с другими генетическими и физическими картами генома.
Основная цель аннотации на уровне белка — присвоить функции белковым продуктам генома. Для этого типа аннотации используются базы данных белковых последовательностей, функциональных доменов и мотивов. Около половины предсказанных белков в новой последовательности генома, как правило, не имеют очевидной функции.
Понимание функции генов и их продуктов в контексте клеточной и организменной физиологии является целью аннотации на уровне процесса. Препятствием для аннотаций на уровне процесса было несоответствие терминов, используемых разными модельными системами. Консорциум Gene Ontology помогает решить эту проблему. [28]
Первое описание комплексной системы аннотаций было опубликовано в 1995 году. [27] Институтом геномных исследований , который впервые выполнил полное секвенирование и анализ генома свободноживущего (несимбиотического ) организма, бактерии Haemophilus influenzae . [27] Система идентифицирует гены, кодирующие все белки, транспортные РНК, рибосомальные РНК, чтобы выполнить первоначальные функциональные назначения. Программа GeneMark , предназначенная для поиска генов, кодирующих белок у Haemophilus influenzae, постоянно меняется и совершенствуется.
Следуя целям, которые проект «Геном человека» оставил после своего закрытия в 2003 году, проект ENCODE был разработан Национальным научно-исследовательским институтом генома человека . Этот проект представляет собой совместный сбор данных о функциональных элементах генома человека, в котором используются технологии секвенирования ДНК нового поколения и массивы геномных плиток, технологии, способные автоматически генерировать большие объемы данных при значительно сниженных затратах на каждую базу, но с теми же точность (ошибка вызова основания) и верность (ошибка сборки).
генов Прогнозирование функций
Хотя аннотация генома в первую очередь основана на сходстве последовательностей (и, следовательно, гомологии ), другие свойства последовательностей могут использоваться для прогнозирования функции генов. Фактически, большинство методов прогнозирования функций генов сосредоточены на белковых последовательностях, поскольку они более информативны и содержат больше возможностей. Например, распределение гидрофобных аминокислот предсказывает наличие трансмембранных сегментов в белках. Однако для прогнозирования функции белка можно также использовать внешнюю информацию, такую как об экспрессии данные генов (или белков), структуру белка или белок-белковые взаимодействия . [29]
биология Вычислительная эволюционная
Эволюционная биология — это изучение происхождения и происхождения видов , а также их изменений с течением времени. Информатика помогла биологам-эволюционистам, позволив исследователям:
- проследить эволюцию большого количества организмов, измеряя изменения в их ДНК , а не только с помощью физической таксономии или физиологических наблюдений,
- сравнивать целые геномы , что позволяет изучать более сложные эволюционные события, такие как дупликация генов , горизонтальный перенос генов и прогнозирование факторов, важных для бактериального видообразования .
- создавать сложные модели вычислительной популяционной генетики для прогнозирования результатов работы системы с течением времени [30]
- отслеживать и обмениваться информацией о все большем количестве видов и организмов
Будущие работы направлены на реконструкцию ныне более сложного древа жизни . [ по мнению кого? ]
Сравнительная геномика [ править ]
Основой сравнительного анализа генома является установление соответствия между генами ( ортологический анализ) или другими геномными признаками у разных организмов. Межгеномные карты создаются для отслеживания эволюционных процессов, ответственных за расхождение двух геномов. Множество эволюционных событий, действующих на различных организационных уровнях, формируют эволюцию генома. На самом низком уровне точковые мутации затрагивают отдельные нуклеотиды. На более высоком уровне крупные хромосомные сегменты подвергаются дупликации, латеральному переносу, инверсии, транспозиции, делеции и вставке. [31] Целые геномы участвуют в процессах гибридизации, полиплоидизации и эндосимбиоза, которые приводят к быстрому видообразованию. Сложность эволюции генома ставит множество интересных задач перед разработчиками математических моделей и алгоритмов, которые прибегают к широкому спектру алгоритмических, статистических и математических методов, начиная от точных, эвристических алгоритмов с фиксированным параметром и алгоритмов аппроксимации для задач, основанных на экономных моделях, до марковских алгоритмов. цепные алгоритмы Монте-Карло для байесовского анализа задач на основе вероятностных моделей.
Многие из этих исследований основаны на обнаружении гомологии последовательностей для отнесения последовательностей к семействам белков . [32]
Пангеномика [ править ]
Пангеномика — это концепция, предложенная в 2005 году Теттелином и Медини. Пангеном — это полный репертуар генов определенной монофилетической таксономической группы. Хотя первоначально оно применялось к близкородственным штаммам вида, его можно применять и к более широкому контексту, например, к роду, типу и т. д. Он разделен на две части: основной геном, набор генов, общий для всех изучаемых геномов (часто гены «домашнего хозяйства», жизненно важные для выживания), а также необязательный/гибкий геном: набор генов, присутствующих не во всех изучаемых геномах, кроме одного или некоторых. Инструмент биоинформатики BPGA можно использовать для характеристики пангенома видов бактерий. [33]
Генетика заболеваний [ править ]
По состоянию на 2013 год существование эффективной высокопроизводительной технологии секвенирования нового поколения позволяет выявлять причины множества различных заболеваний человека. Простое менделевское наследование наблюдалось для более чем 3000 расстройств, которые были идентифицированы в онлайн-базе данных «Менделевское наследование у человека» , но сложные заболевания представляют собой более сложную задачу. Исследования ассоциаций обнаружили множество отдельных генетических областей, которые по отдельности слабо связаны со сложными заболеваниями (такими как бесплодие , [34] рак молочной железы [35] и болезнь Альцгеймера [36] ), а не одна причина. [37] [38] В настоящее время существует множество проблем с использованием генов для диагностики и лечения, например, то, что мы не знаем, какие гены важны, или насколько стабильный выбор обеспечивает алгоритм. [39]
Полногеномные исследования ассоциаций успешно выявили тысячи общих генетических вариантов сложных заболеваний и признаков; однако эти общие варианты объясняют лишь небольшую часть наследственности. [40] Редкие варианты могут частично объяснить недостающую наследственность . [41] Крупномасштабные исследования секвенирования всего генома быстро секвенировали миллионы целых геномов, и такие исследования выявили сотни миллионов редких вариантов . [42] Функциональные аннотации предсказывают эффект или функцию генетического варианта и помогают определить приоритет редких функциональных вариантов, а включение этих аннотаций может эффективно повысить эффективность генетической ассоциации анализа редких вариантов в исследованиях полногеномного секвенирования. [43] Некоторые инструменты были разработаны для обеспечения комплексного анализа ассоциаций редких вариантов для данных полногеномного секвенирования, включая интеграцию данных генотипа и их функциональных аннотаций, анализ ассоциаций, сводку результатов и визуализацию. [44] [45] Метаанализ исследований полногеномного секвенирования обеспечивает привлекательное решение проблемы сбора больших объемов выборок для обнаружения редких вариантов, связанных со сложными фенотипами. [46]
Анализ мутаций при раке [ править ]
При раке геномы пораженных клеток перестраиваются сложным или непредсказуемым образом. В дополнение к массивам однонуклеотидного полиморфизма , выявляющим точечные мутации , вызывающие рак, олигонуклеотидные микрочипы могут использоваться для выявления хромосомных приобретений и потерь (так называемая сравнительная геномная гибридизация ). Эти методы обнаружения генерируют терабайты данных за эксперимент. Часто обнаруживается, что данные содержат значительную изменчивость или шум , и поэтому скрытая модель Маркова разрабатываются и методы анализа точек изменений, чтобы сделать вывод о реальных изменениях количества копий . [ нужна ссылка ]
можно использовать два важных принципа Для идентификации рака по мутациям в экзоме . Во-первых, рак — это заболевание, вызванное накопленными соматическими мутациями в генах. Во-вторых, рак содержит мутации водителя, которые необходимо отличать от мутаций пассажиров. [47]
Дальнейшие улучшения в биоинформатике могут позволить классифицировать типы рака путем анализа мутаций в геноме, вызванных раком. Более того, отслеживание пациентов по мере прогрессирования заболевания может стать возможным в будущем с помощью последовательности образцов рака. Другой тип данных, требующий развития новой информатики, — это анализ поражений, которые оказались рецидивирующими среди многих опухолей. [48]
генов Экспрессия белков и
генов экспрессии Анализ
Экспрессию секвенирование многих генов можно определить путем измерения уровней мРНК с помощью нескольких методов, включая микрочипы , секвенирование экспрессированных последовательностей кДНК (EST), меток последовательного анализа экспрессии генов (SAGE), массово-параллельное сигнатурное секвенирование (MPSS), RNA-Seq , также известный как «целое транскриптомное секвенирование» (WTSS), или различные применения мультиплексной гибридизации in-situ. Все эти методы чрезвычайно подвержены шуму и/или предвзятости при биологических измерениях, а основная область исследований в вычислительной биологии включает разработку статистических инструментов для отделения сигнала от шума в высокопроизводительных исследованиях экспрессии генов. [49] Такие исследования часто используются для определения генов, участвующих в заболевании: можно сравнить данные микрочипов раковых эпителиальных клеток с данными нераковых клеток, чтобы определить транскрипты, активация которых повышена или понижена в определенной популяции раковых клеток. .

Анализ экспрессии белка
Белковые микроматрицы и масс-спектрометрия (МС) с высокой пропускной способностью (HT) могут предоставить моментальный снимок белков, присутствующих в биологическом образце. Первый подход сталкивается с теми же проблемами, что и микрочипы, нацеленные на мРНК, второй включает в себя проблему сопоставления больших объемов данных о массах с предсказанными массами из баз данных последовательностей белков, а также сложный статистический анализ образцов при обнаружении нескольких неполных пептидов из каждого белка. Локализация клеточного белка в тканевом контексте может быть достигнута с помощью аффинной протеомики, отображаемой в виде пространственных данных, основанных на иммуногистохимии и тканевых микрочипах . [50]
Анализ регулирования [ править ]
Регуляция генов — это сложный процесс, при котором сигнал, например внеклеточный сигнал, такой как гормон , в конечном итоге приводит к увеличению или снижению активности одного или нескольких белков . Методы биоинформатики применялись для изучения различных этапов этого процесса.
Например, экспрессия генов может регулироваться близлежащими элементами генома. Анализ промотора включает идентификацию и изучение мотивов последовательности в ДНК, окружающей белок-кодирующую область гена. Эти мотивы влияют на степень, в которой эта область транскрибируется в мРНК. Элементы- энхансеры , расположенные далеко от промотора, также могут регулировать экспрессию генов посредством трехмерных петлевых взаимодействий. Эти взаимодействия могут быть определены с помощью биоинформатического анализа экспериментов по захвату конформации хромосом .
Данные об экспрессии можно использовать для вывода о регуляции генов: можно сравнить данные микрочипов из самых разных состояний организма, чтобы сформировать гипотезы о генах, участвующих в каждом состоянии. В одноклеточном организме можно сравнить стадии клеточного цикла , а также различные стрессовые состояния (тепловой шок, голодание и т. д.). Затем к данным экспрессии можно применить алгоритмы кластеризации, чтобы определить, какие гены экспрессируются совместно. Например, в вышележащих регионах (промоторах) совместно экспрессируемых генов можно искать чрезмерно представленные регуляторные элементы . Примерами алгоритмов кластеризации, применяемых при кластеризации генов, являются кластеризация k-средних , самоорганизующиеся карты (SOM), иерархическая кластеризация и консенсусной кластеризации методы .
Анализ клеточной организации [ править ]
Было разработано несколько подходов для анализа расположения органелл, генов, белков и других компонентов внутри клеток. Категория онтологии генов , клеточный компонент , была разработана для определения субклеточной локализации во многих биологических базах данных .
и Микроскопия изображений анализ
Микроскопические изображения позволяют определить местонахождение органелл , а также молекул, которые могут быть источником аномалий при заболеваниях.
Локализация белка [ править ]
Обнаружение местоположения белков позволяет нам предсказать, что они делают. Это называется предсказанием функции белка . Например, если белок обнаружен в ядре , он может участвовать в регуляции генов или сплайсинге . Напротив, если белок обнаружен в митохондриях , он может участвовать в дыхании или других метаболических процессах . Существуют хорошо развитые ресурсы для прогнозирования субклеточной локализации белков , включая базы данных субклеточной локализации белков и инструменты прогнозирования. [51] [52]
Ядерная организация хроматина [ править ]
Данные высокопроизводительных экспериментов по захвату конформации хромосом , таких как Hi-C (эксперимент) ChIA -PET , могут предоставить информацию о трехмерной структуре и ядерной организации хроматина и . Биоинформационные задачи в этой области включают разделение генома на домены, такие как топологически ассоциированные домены (TAD), которые организованы вместе в трехмерном пространстве. [53]
Структурная биоинформатика [ править ]

Поиск структуры белков является важным применением биоинформатики. Критическая оценка прогнозирования структуры белка (CASP) — это открытый конкурс, в ходе которого исследовательские группы со всего мира представляют модели белков для оценки неизвестных моделей белков. [54] [55]
Аминокислотная последовательность [ править ]
Линейная аминокислотная последовательность белка называется первичной структурой . Первичную структуру можно легко определить по последовательности кодонов гена ДНК, который ее кодирует. В большинстве белков первичная структура однозначно определяет трехмерную структуру белка в его нативной среде. Исключением является неправильно свернутый белок, участвующий в губчатой энцефалопатии крупного рогатого скота . Эта структура связана с функцией белка. Дополнительная структурная информация включает вторичную , третичную и четвертичную структуру. Жизнеспособное общее решение для предсказания функции белка остается открытой проблемой. До сих пор большинство усилий было направлено на эвристику, которая работает большую часть времени. [ нужна ссылка ]
Гомология [ править ]
В геномной отрасли биоинформатики гомология используется для предсказания функции гена: если последовательность гена A , функция которого известна, гомологична последовательности гена B, функция которого неизвестна, можно сделать вывод, что B может разделить функцию А. В структурной биоинформатике гомология используется для определения того, какие части белка важны для формирования структуры и взаимодействия с другими белками. Моделирование гомологии используется для прогнозирования структуры неизвестного белка на основе существующих гомологичных белков.
Одним из примеров этого является гемоглобин человека и гемоглобин бобовых ( леггемоглобин ), которые являются дальними родственниками одного и того же суперсемейства белков . Оба служат одной и той же цели – транспортировке кислорода в организме. Хотя оба этих белка имеют совершенно разные аминокислотные последовательности, их белковые структуры практически идентичны, что отражает их почти идентичные цели и общего предка. [56]
Другие методы прогнозирования структуры белка включают в себя создание нитей белка и de novo моделирование на основе физики (с нуля).
Другой аспект структурной биоинформатики включает использование белковых структур для моделей виртуального скрининга , таких как модели количественной взаимосвязи структура-активность и протеохемометрические модели (PCM). Кроме того, кристаллическую структуру белка можно использовать при моделировании, например, исследований связывания лигандов и в исследованиях кремнеземного мутагенеза.
Программное обеспечение 2021 года глубокого обучения на основе алгоритмов под названием AlphaFold , разработанное Google DeepMind , значительно превосходит все другие методы программного обеспечения для прогнозирования. [57] [ как? ] и опубликовал предсказанные структуры для сотен миллионов белков в базе данных структур белков AlphaFold. [58]
Сетевая биология системная и
Сетевой анализ направлен на понимание взаимоотношений внутри биологических сетей, таких как метаболические сети или сети межбелкового взаимодействия . Хотя биологические сети могут быть построены из одного типа молекул или объектов (например, генов), сетевая биология часто пытается интегрировать множество различных типов данных, таких как белки, небольшие молекулы, данные об экспрессии генов и другие, которые все связаны физически. , функционально или и то, и другое.
Системная биология предполагает использование компьютерного моделирования клеточных пути подсистем (таких как сети метаболитов и ферментов , которые включают метаболизм , передачи сигналов и сети регуляции генов ) для анализа и визуализации сложных связей этих клеточных процессов. Искусственная жизнь или виртуальная эволюция пытается понять эволюционные процессы посредством компьютерного моделирования простых (искусственных) форм жизни.
Сети молекулярного взаимодействия

Десятки тысяч трехмерных белковых структур были определены с помощью рентгеновской кристаллографии и спектроскопии ядерного магнитного резонанса белков (ЯМР белков), и центральный вопрос структурной биоинформатики заключается в том, практично ли прогнозировать возможные межбелковые взаимодействия только на основе этих структур. 3D-формы без проведения экспериментов по межбелковому взаимодействию . было разработано множество методов Для решения проблемы докинга белков , хотя кажется, что в этой области еще предстоит проделать большую работу.
Другие взаимодействия, встречающиеся в этой области, включают белок-лиганд (включая лекарственное средство) и белок-пептид . Молекулярно-динамическое моделирование движения атомов вокруг вращающихся связей является фундаментальным принципом вычислительных алгоритмов , называемых алгоритмами стыковки, для изучения молекулярных взаимодействий .
Информатика биоразнообразия
Информатика биоразнообразия занимается сбором и анализом данных о биоразнообразии , таких как таксономические базы данных или микробиома данные . Примеры такого анализа включают филогенетику , моделирование ниш , видового богатства картирование , штрих-кодирование ДНК или инструменты идентификации видов . Растущей областью также является макроэкология , то есть изучение того, как биоразнообразие связано с экологией и воздействием человека, например, изменением климата .
Другие [ править ]
Литературный анализ [ править ]
Огромное количество опубликованной литературы делает практически невозможным для отдельных лиц читать каждую статью, что приводит к разрознению подобластей исследований. Целью анализа литературы является использование компьютерной и статистической лингвистики для изучения этой растущей библиотеки текстовых ресурсов. Например:
- Распознавание сокращений – определение полной формы и сокращений биологических терминов.
- Распознавание именованного объекта - распознавание биологических терминов, таких как имена генов.
- Белково-белковое взаимодействие - определите, какие белки с какими белками взаимодействуют из текста.
Область исследований опирается на статистику и компьютерную лингвистику .
Высокопроизводительный изображений анализ
Вычислительные технологии используются для автоматизации обработки, количественной оценки и анализа больших объемов биомедицинских изображений с высоким содержанием информации . Современные системы анализа изображений наблюдения наблюдателя могут повысить точность , объективность и скорость . Анализ изображений важен как для диагностики , так и для исследований. Некоторые примеры:
- высокопроизводительная и высокоточная количественная оценка и субклеточная локализация ( скрининг высокого содержания , цитогистопатология, информатика биоизображений )
- морфометрия
- анализ и визуализация клинических изображений
- определение в реальном времени режимов потока воздуха в дыхательных легких живых животных
- количественная оценка размера окклюзии в режиме реального времени на основе изображений развития и восстановления во время артериального повреждения
- проведение поведенческих наблюдений на основе расширенных видеозаписей лабораторных животных
- инфракрасные измерения для определения метаболической активности
- вывод о перекрытии клонов при картировании ДНК , например, по шкале Салстона
ячеек Высокопроизводительный анализ данных отдельных
Вычислительные методы используются для анализа высокопроизводительных и малообъемных данных об отдельных клетках, например, полученных с помощью проточной цитометрии . Эти методы обычно включают поиск популяций клеток, соответствующих конкретному болезненному состоянию или экспериментальному состоянию.
и Онтологии интеграция данных
Биологические онтологии представляют собой ориентированные ациклические графы словарей управляемых . Они создают категории для биологических концепций и описаний, чтобы их можно было легко анализировать с помощью компьютеров. При такой классификации можно получить дополнительную выгоду от целостного и интегрированного анализа. [ нужна ссылка ]
OBO Foundry была попыткой стандартизировать определенные онтологии. Одной из наиболее распространенных является онтология генов , описывающая функции генов. Существуют также онтологии, описывающие фенотипы.
Базы данных [ править ]
Базы данных необходимы для исследований и приложений в области биоинформатики. Базы данных существуют для множества различных типов информации, включая последовательности ДНК и белков, молекулярные структуры, фенотипы и биоразнообразие. Базы данных могут содержать как эмпирические данные (полученные непосредственно в результате экспериментов), так и прогнозируемые данные (полученные в результате анализа существующих данных). Они могут быть специфичными для конкретного организма, пути или молекулы, представляющей интерес. Альтернативно они могут включать данные, собранные из нескольких других баз данных. Базы данных могут иметь разные форматы, механизмы доступа и быть общедоступными или частными.
Ниже перечислены некоторые из наиболее часто используемых баз данных:
- Используется в анализе биологических последовательностей: Genbank , UniProt.
- Используется в структурном анализе: Банк данных белков (PDB).
- Используется для поиска семейств белков и поиска мотивов : InterPro , Pfam.
- Используется для секвенирования следующего поколения: архив чтения последовательности
- Используется в сетевом анализе: базы данных метаболических путей ( KEGG , BioCyc ), базы данных анализа взаимодействия, функциональные сети.
- Используется при проектировании синтетических генетических схем: GenoCAD. [ нужна ссылка ]
Программное обеспечение и инструменты [ править ]
Программные инструменты для биоинформатики включают простые инструменты командной строки, более сложные графические программы и автономные веб-сервисы. Их производят биоинформационные компании или государственные учреждения.
с открытым исходным кодом Программное обеспечение для биоинформатики
Многие бесплатные программные инструменты с открытым исходным кодом существовали и продолжали расти с 1980-х годов. [60] Сочетание постоянной потребности в новых алгоритмах для анализа новых типов биологических данных, потенциала инновационных экспериментов in silico и свободно доступных баз открытого кода создало для исследовательских групп возможности внести свой вклад в обе биоинформатики независимо от финансирования . Инструменты с открытым исходным кодом часто выступают в качестве инкубаторов идей или поддерживаемых сообществом плагинов для коммерческих приложений. Они также могут предоставлять стандарты де-факто и общие объектные модели для решения проблемы интеграции биоинформации.
Программное обеспечение для биоинформатики с открытым исходным кодом включает Bioconductor , BioPerl , Biopython , BioJava , BioJS , BioRuby , Bioclipse , EMBOSS , .NET Bio, Orange с надстройкой для биоинформатики, Apache Taverna , UGENE и GenoCAD .
Некоммерческий Фонд Открытой Биоинформатики [60] а ежегодная конференция по биоинформатике с открытым исходным кодом продвигает программное обеспечение для биоинформатики с открытым исходным кодом. [61]
Веб-сервисы в биоинформатике [ править ]
Интерфейсы на основе SOAP и REST были разработаны, чтобы позволить клиентским компьютерам использовать алгоритмы, данные и вычислительные ресурсы с серверов в других частях мира. Основное преимущество заключается в том, что конечным пользователям не придется иметь дело с накладными расходами на обслуживание программного обеспечения и баз данных.
Основные биоинформатические услуги классифицируются EBI на три категории: SSS (службы поиска последовательностей), MSA (множественное выравнивание последовательностей) и BSA (анализ биологических последовательностей). [62] Доступность этих сервисно-ориентированных биоинформатических ресурсов демонстрирует применимость сетевых биоинформатических решений и варьируется от набора автономных инструментов с общим форматом данных под единым веб-интерфейсом до интегративных, распределенных и расширяемых систем управления рабочими процессами в области биоинформатики. .
Системы управления биоинформатики рабочими процессами
Система управления рабочими процессами биоинформатики — это специализированная форма системы управления рабочими процессами, разработанная специально для составления и выполнения серии шагов вычислений или обработки данных или рабочего процесса в приложении биоинформатики. Такие системы предназначены для
- предоставить простую в использовании среду для отдельных специалистов по приложениям, позволяющих им создавать свои собственные рабочие процессы,
- предоставить ученым интерактивные инструменты, позволяющие им выполнять рабочие процессы и просматривать результаты в режиме реального времени,
- упростить процесс совместного использования и повторного использования рабочих процессов между учеными, а также
- позволяют ученым отслеживать происхождение результатов выполнения рабочего процесса и этапов его создания.
Некоторые платформы, предоставляющие эту услугу: Galaxy , Kepler , Taverna , UGENE , Anduril , HIVE .
и биокомпьютеров объекты Биокомпьюты
В 2014 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США спонсировало конференцию, проведенную в кампусе Национального института здравоохранения в Бетесде, чтобы обсудить воспроизводимость в биоинформатике. [63] В течение следующих трех лет консорциум заинтересованных сторон регулярно встречался, чтобы обсудить то, что впоследствии станет парадигмой биокомпьютеров. [64] В число этих заинтересованных сторон входили представители правительства, промышленности и академических организаций. Лидеры сессии представляли многочисленные филиалы институтов и центров FDA и NIH, некоммерческие организации, включая Human Variome Project и Европейскую федерацию медицинской информатики , а также исследовательские институты, включая Стэнфорд , Нью-Йоркский центр генома и Университет Джорджа Вашингтона .
Было решено, что парадигма BioCompute будет иметь форму цифровых «лабораторных ноутбуков», которые позволят воспроизводить, воспроизводить, анализировать и повторно использовать протоколы биоинформатики. Это было предложено для обеспечения большей преемственности внутри исследовательской группы в ходе обычного потока кадров, одновременно способствуя обмену идеями между группами. FDA США профинансировало эту работу, чтобы информация о трубопроводах была более прозрачной и доступной для сотрудников регулирующих органов. [65]
В 2016 году группа вновь собралась в НИЗ в Бетесде и обсудила потенциал объекта BioCompute — примера парадигмы BioCompute. Эта работа была скопирована как документ «стандартного пробного использования», так и как препринт, загруженный в bioRxiv. Объект BioCompute позволяет делиться записью в формате JSON между сотрудниками, сотрудниками и регулирующими органами. [66] [67]
Образовательные платформы [ править ]
Биоинформатика во многих университетах преподается не только в магистратуре . Вычислительная природа биоинформатики позволяет использовать ее для компьютерного и онлайн-обучения . [68] [69] Программные платформы, предназначенные для обучения концепциям и методам биоинформатики, включают Rosalind и онлайн-курсы, предлагаемые через учебный портал Швейцарского института биоинформатики . Канадские семинары по биоинформатике размещают на своем веб-сайте видео и слайды с учебных семинаров по лицензии Creative Commons . Проект 4273π или проект 4273pi [70] также предлагает бесплатные образовательные материалы с открытым исходным кодом. Курс проводится на недорогих компьютерах Raspberry Pi и используется для обучения взрослых и школьников. [71] [72] 4273 активно разрабатывается консорциумом ученых и исследователей, которые проводили исследования в области биоинформатики с использованием компьютеров Raspberry Pi и операционной системы 4273π. [73] [74]
Платформы MOOC также предоставляют онлайн-сертификацию по биоинформатике и смежным дисциплинам, включая специализацию Coursera по биоинформатике ( Калифорнийский университет в Сан-Диего ) и специализацию по науке о геномных данных ( Джонс Хопкинс ), а также Гарвард по анализу данных EdX для наук о жизни XSeries ( ) .
Конференции [ править ]
Есть несколько крупных конференций, посвященных биоинформатике. Некоторые из наиболее ярких примеров — «Интеллектуальные системы для молекулярной биологии» (ISMB), Европейская конференция по вычислительной биологии (ECCB) и «Исследования в области вычислительной молекулярной биологии» (RECOMB).
См. также [ править ]
- Информатика биоразнообразия
- Биоинформатические компании
- Вычислительная биология
- Компьютерное биомоделирование
- Вычислительная геномика
- Кибербиобезопасность
- Функциональная геномика
- База данных генных заболеваний
- Информатика здравоохранения
- Международное общество вычислительной биологии
- Прыгающая библиотека
- Список учреждений биоинформатики
- Список программного обеспечения для биоинформатики с открытым исходным кодом
- Список журналов по биоинформатике
- Метаболомика
- Последовательность нуклеиновой кислоты
- Филогенетика
- Протеомика
Ссылки [ править ]
- ^ Ганюк П. (17 августа 2021 г.). Алгоритмы в биоинформатике: теория и реализация (1-е изд.). Уайли. стр. 1–528. ISBN 978-1-119-69796-1 .
- ^ Леск А.М. (26 июля 2013 г.). «Биоинформатика» . Британская энциклопедия . Архивировано из оригинала 14 апреля 2021 года . Проверено 17 апреля 2017 г.
- ^ Jump up to: Перейти обратно: а б Сим А.Ю., Минари П., Левитт М. (июнь 2012 г.). «Моделирование нуклеиновых кислот» . Современное мнение в области структурной биологии . 22 (3): 273–8. дои : 10.1016/j.sbi.2012.03.012 . ПМК 4028509 . ПМИД 22538125 .
- ^ Доусон В.К., Мачейчик М., Янковска Э.Дж., Буйницки Дж.М. (июль 2016 г.). «Грубозернистое моделирование 3D-структуры РНК» . методы . 103 :138–56. дои : 10.1016/j.ymeth.2016.04.026 . ПМИД 27125734 .
- ^ Кмиецик С., Гронт Д., Колински М., Витеска Л., Давид А.Е., Колински А. (июль 2016 г.). «Крупнозернистые белковые модели и их применение» . Химические обзоры . 116 (14): 7898–936. doi : 10.1021/acs.chemrev.6b00163 . ПМИД 27333362 .
- ^ Вонг КС (2016). Вычислительная биология и биоинформатика: регуляция генов . CRC Press/Taylor & Francisco Group. ISBN 978-1-4987-2497-5 .
- ^ Джойс А.П., Чжан С., Брэдли П., Хавранек Дж.Дж. (январь 2015 г.). «Структурное моделирование белка: специфичность ДНК» . Брифинги по функциональной геномике . 14 (1): 39–49. дои : 10.1093/bfgp/elu044 . ПМЦ 4366589 . ПМИД 25414269 .
- ^ Спига Э., Дегиакоми М.Т., Даль Пераро М. (2014). «Новые стратегии интегративного динамического моделирования макромолекулярной сборки». В Карабенчевой-Кристовой Т. (ред.). Биомолекулярное моделирование и моделирование . Достижения в области химии белков и структурной биологии. Том. 96. Академическая пресса. стр. 77–111. дои : 10.1016/bs.apcsb.2014.06.008 . ISBN 978-0-12-800013-7 . ПМИД 25443955 .
- ^ Циемни М., Курчински М., Камель К., Колински А., Алам Н., Шулер-Фурман О. и др. (август 2018 г.). «Белко-пептидный докинг: возможности и проблемы» . Открытие наркотиков сегодня . 23 (8): 1530–1537. дои : 10.1016/j.drudis.2018.05.006 . ПМИД 29733895 .
- ^ Узунис, Калифорния, Валенсия А (2003). «Ранняя биоинформатика: рождение дисциплины — личный взгляд» . Биоинформатика . 19 (17): 2176–2190. doi : 10.1093/биоинформатика/btg309 . ПМИД 14630646 .
- ^ Jump up to: Перейти обратно: а б Хогевег П. (2011). «Корни биоинформатики в теоретической биологии» . PLOS Вычислительная биология . 7 (3): e1002021. Бибкод : 2011PLSCB...7E2021H . дои : 10.1371/journal.pcbi.1002021 . ПМК 3068925 . ПМИД 21483479 .
- ^ Хеспер Б., Хогевег П. (1970). «БИОИНФОРМАТИКА: рабочая концепция». Хамелеон (на голландском языке). 1 (6): 28–29.
- ^ Хеспер Б., Хогевег П. (2021). «Биоинформатика: рабочая концепция. Перевод книги Б. Хеспера и П. Хогевега «Биоинформатика: рабочая концепция». arXiv : 2111.11832v1 [ q-bio.OT ].
- ^ Хогевег П. (1978). «Моделирование роста клеточных форм». Моделирование . 31 (3): 90–96. дои : 10.1177/003754977803100305 . S2CID 61206099 .
- ^ Колби Б. (2022). «Стоимость секвенирования полного генома» . Sequencing.com . Архивировано из оригинала 15 марта 2022 года . Проверено 8 апреля 2022 г.
- ^ Сэнгер Ф, Таппи Х (1951). «Аминокислотная последовательность фенилаланильной цепи инсулина. I. Идентификация низших пептидов из частичных гидролизатов» . Биохимический журнал . 49 (4): 463–81. дои : 10.1042/bj0490463 . ПМК 1197535 . ПМИД 14886310 .
- ^ Сэнгер Ф., Томпсон Э.О. (1953). «Аминокислотная последовательность в гликолевой цепи инсулина. I. Идентификация низших пептидов из частичных гидролизатов» . Биохимический журнал . 53 (3): 353–66. дои : 10.1042/bj0530353 . ПМК 1198157 . ПМИД 13032078 .
- ^ Муди Дж. (2004). Цифровой код жизни: как биоинформатика совершает революцию в науке, медицине и бизнесе . Хобокен, Нью-Джерси, США: John Wiley & Sons. ISBN 978-0-471-32788-2 .
- ^ Дайхофф М.О., Эк Р.В., Чанг М.А., Сочард М.Р. (1965). АТЛАС ПОСЛЕДОВАТЕЛЬНОСТИ И СТРУКТУРЫ БЕЛКОВ (PDF) . Силвер Спринг, Мэриленд, США: Национальный фонд биомедицинских исследований. LCCN 65-29342 .
- ^ Эк Р.В., Дайхофф, Миссури (апрель 1966 г.). «Эволюция структуры ферредоксина на основе живых остатков примитивных аминокислотных последовательностей». Наука . 152 (3720): 363–6. Бибкод : 1966Sci...152..363E . дои : 10.1126/science.152.3720.363 . ПМИД 17775169 . S2CID 23208558 .
- ^ Джонсон Дж., Ву Т.Т. (январь 2000 г.). «База данных Кабата и ее приложения: 30 лет после первого графика изменчивости» . Исследования нуклеиновых кислот . 28 (1): 214–8. дои : 10.1093/нар/28.1.214 . ПМЦ 102431 . ПМИД 10592229 .
- ^ Эриксон Дж.В., Альтман Г.Г. (1979). «Поиск закономерностей в нуклеотидной последовательности генома MS2». Журнал математической биологии . 7 (3): 219–230. дои : 10.1007/BF00275725 . S2CID 85199492 .
- ^ Шульман М.Дж., Стейнберг К.М., Уэстморленд Н. (февраль 1981 г.). «Кодирующая функция нуклеотидных последовательностей может быть определена с помощью статистического анализа». Журнал теоретической биологии . 88 (3): 409–20. Бибкод : 1981JThBi..88..409S . дои : 10.1016/0022-5193(81)90274-5 . ПМИД 6456380 .
- ^ Сюн Дж (2006). Основная биоинформатика . Кембридж, Соединенное Королевство: Издательство Кембриджского университета. стр. 4 . ISBN 978-0-511-16815-4 – через Интернет-архив.
- ^ Сэнгер Ф., Air GM, Баррелл Б.Г., Браун Н.Л., Коулсон А.Р., Фиддес К.А. и др. (февраль 1977 г.). «Нуклеотидная последовательность ДНК бактериофага phi X174». Природа . 265 (5596): 687–95. Бибкод : 1977Natur.265..687S . дои : 10.1038/265687a0 . ПМИД 870828 . S2CID 4206886 .
- ^ Бенсон Д.А., Карш-Мизрачи И., Липман Д.Д., Остелл Дж., Уилер Д.Л. (январь 2008 г.). «ГенБанк» . Исследования нуклеиновых кислот . 36 (Проблема с базой данных): D25-30. дои : 10.1093/нар/gkm929 . ПМК 2238942 . ПМИД 18073190 .
- ^ Jump up to: Перейти обратно: а б с Флейшманн Р.Д., Адамс М.Д., Уайт О., Клейтон Р.А., Киркнесс Э.Ф., Керлаваж А.Р. и др. (июль 1995 г.). «Полногеномное случайное секвенирование и сборка Haemophilus influenzae Rd». Наука . 269 (5223): 496–512. Бибкод : 1995Sci...269..496F . дои : 10.1126/science.7542800 . ПМИД 7542800 .
- ^ Штейн Л. (2001). «Аннотация генома: от последовательности к биологии». Природа . 2 (7): 493–503. дои : 10.1038/35080529 . ПМИД 11433356 . S2CID 12044602 .
- ^ Эрдин С., Лисевски А.М., Лихтарге О. (апрель 2011 г.). «Прогнозирование функции белка: к интеграции показателей сходства» . Современное мнение в области структурной биологии . 21 (2): 180–8. дои : 10.1016/j.sbi.2011.02.001 . ПМК 3120633 . ПМИД 21353529 .
- ^ Карвахаль-Родригес А. (март 2010 г.). «Моделирование генов и геномов вперед во времени» . Современная геномика . 11 (1): 58–61. дои : 10.2174/138920210790218007 . ПМЦ 2851118 . ПМИД 20808525 .
- ^ Браун Т.А. (2002). «Мутация, репарация и рекомбинация». Геномы (2-е изд.). Манчестер (Великобритания): Оксфорд.
- ^ Картер Н.П., Фиглер Х., Пайпер Дж. (октябрь 2002 г.). «Сравнительный анализ технологий сравнительной геномной гибридизации на микрочипах: отчет о семинаре, спонсируемом Wellcome Trust». Цитометрия . 49 (2): 43–8. дои : 10.1002/cyto.10153 . ПМИД 12357458 .
- ^ Чаудхари Н.М., Гупта В.К., Дутта С. (апрель 2016 г.). «BPGA — сверхбыстрый конвейер пангеномного анализа» . Научные отчеты . 6 : 24373. Бибкод : 2016NatSR...624373C . дои : 10.1038/srep24373 . ПМЦ 4829868 . ПМИД 27071527 .
- ^ Астон КИ (май 2014 г.). «Генетическая предрасположенность к мужскому бесплодию: новости полногеномных ассоциативных исследований» . Андрология . 2 (3): 315–21. дои : 10.1111/j.2047-2927.2014.00188.x . ПМИД 24574159 . S2CID 206007180 .
- ^ Верон А., Блейн С., Кокс Д.Г. (2014). «Полногеномные ассоциативные исследования и клиника: в центре внимания рак молочной железы». Биомаркеры в медицине . 8 (2): 287–96. дои : 10.2217/bmm.13.121 . ПМИД 24521025 .
- ^ Тосто Дж., Райц К. (октябрь 2013 г.). «Полногеномные исследования ассоциации при болезни Альцгеймера: обзор» . Текущие отчеты по неврологии и нейробиологии . 13 (10): 381. дои : 10.1007/s11910-013-0381-0 . ПМЦ 3809844 . ПМИД 23954969 .
- ^ Лондин Э., Ядав П., Суррей С., Крика Л.Дж., Фортина П. (2013). «Использование анализа сцепления, общегеномных исследований ассоциаций и секвенирования следующего поколения для выявления мутаций, вызывающих заболевания». Фармакогеномика . Методы молекулярной биологии. Том. 1015. стр. 127–46. дои : 10.1007/978-1-62703-435-7_8 . ISBN 978-1-62703-434-0 . ПМИД 23824853 .
- ^ Хиндорфф Л.А., Сетупати П., Джанкинс Х.А., Рамос Э.М., Мехта Дж.П., Коллинз Ф.С. и др. (июнь 2009 г.). «Потенциальные этиологические и функциональные последствия полногеномных ассоциативных локусов для болезней и черт человека» . Труды Национальной академии наук Соединенных Штатов Америки . 106 (23): 9362–7. Бибкод : 2009PNAS..106.9362H . дои : 10.1073/pnas.0903103106 . ПМЦ 2687147 . ПМИД 19474294 .
- ^ Зал ЛО (2010). «Поиск правильных генов для прогнозирования заболеваний и прогнозов». 2010 Международная конференция по системным наукам и инженерии . стр. 1–2. дои : 10.1109/ICSSE.2010.5551766 . ISBN 978-1-4244-6472-2 . S2CID 21622726 .
- ^ Манолио Т.А., Коллинз Ф.С., Кокс Н.Дж., Гольдштейн Д.Б., Хиндорфф Л.А., Хантер DJ и др. (октябрь 2009 г.). «Обнаружение недостающей наследственности сложных заболеваний» . Природа . 461 (7265): 747–753. Бибкод : 2009Natur.461..747M . дои : 10.1038/nature08494 . ПМЦ 2831613 . ПМИД 19812666 .
- ^ Вайнштейн П., Джайн Д., Чжэн З., Аслибекян С., Беккер Д., Би В. и др. (март 2022 г.). «Оценка вклада редких вариантов в наследственность сложных признаков на основе данных полногеномной последовательности» . Природная генетика . 54 (3): 263–273. дои : 10.1038/s41588-021-00997-7 . ПМЦ 9119698 . ПМИД 35256806 .
- ^ Талиун Д., Харрис Д.Н., Кесслер М.Д., Карлсон Дж., Шпих З.А., Торрес Р. и др. (февраль 2021 г.). «Секвенирование 53 831 разнообразного генома из программы NHLBI TOPMed» . Природа . 590 (7845): 290–299. Бибкод : 2021Natur.590..290T . дои : 10.1038/s41586-021-03205-y . ПМЦ 7875770 . ПМИД 33568819 .
- ^ Ли Х, Ли З, Чжоу Х, Гейнор С.М., Лю Ю, Чен Х и др. (сентябрь 2020 г.). «Динамическое включение нескольких функциональных аннотаций in silico расширяет возможности анализа ассоциаций редких вариантов в крупных масштабных исследованиях полногеномного секвенирования» . Природная генетика . 52 (9): 969–983. дои : 10.1038/s41588-020-0676-4 . ПМЦ 7483769 . ПМИД 32839606 .
- ^ Ли З., Ли Икс, Чжоу Х., Гейнор С.М., Сельварадж М.С., Арапоглу Т. и др. (декабрь 2022 г.). «Система обнаружения некодирующих ассоциаций редких вариантов в крупномасштабных исследованиях полногеномного секвенирования» . Природные методы . 19 (12): 1599–1611. дои : 10.1038/s41592-022-01640-x . ПМЦ 10008172 . ПМИД 36303018 . S2CID 243873361 .
- ^ «STAARpipeline: универсальный редкий вариант инструмента для данных полногеномного секвенирования в масштабе биобанка». Природные методы . 19 (12): 1532–1533. Декабрь 2022 г. doi : 10.1038/s41592-022-01641-w . ПМИД 36316564 . S2CID 253246835 .
- ^ Ли Х, Квик С., Чжоу Х., Гейнор С.М., Лю Ю., Чен Х. и др. (январь 2023 г.). «Мощный, масштабируемый и ресурсоэффективный метаанализ ассоциаций редких вариантов в крупных исследованиях полногеномного секвенирования» . Природная генетика . 55 (1): 154–164. дои : 10.1038/s41588-022-01225-6 . ПМЦ 10084891 . ПМИД 36564505 . S2CID 255084231 .
- ^ Васкес М., де ла Торре В., Валенсия А (27 декабря 2012 г.). «Глава 14: Анализ генома рака» . PLOS Вычислительная биология . 8 (12): e1002824. Бибкод : 2012PLSCB...8E2824V . дои : 10.1371/journal.pcbi.1002824 . ПМЦ 3531315 . ПМИД 23300415 .
- ^ Хе-Юнг ЕС, Джасвиндер К., Мартин К., Сэмюэл А.А., Марко А.М. (2014). «Секвенирование второго поколения для анализа генома рака». В Деллере Дж., Бермане Дж.Н., Арчечи Р.Дж. (ред.). Геномика рака . Бостон (США): Академическая пресса. стр. 13–30. дои : 10.1016/B978-0-12-396967-5.00002-5 . ISBN 978-0-12-396967-5 .
- ^ Грау Дж, Бен-Гал I, Пош С, Гросс I (июль 2006 г.). «VOMBAT: прогнозирование сайтов связывания транскрипционных факторов с использованием байесовских деревьев переменного порядка» . Исследования нуклеиновых кислот . 34 (проблема с веб-сервером): W529-33. дои : 10.1093/нар/gkl212 . ПМЦ 1538886 . ПМИД 16845064 .
- ^ «Атлас белков человека» . www.proteinatlas.org . Архивировано из оригинала 4 марта 2020 года . Проверено 2 октября 2017 г.
- ^ «Человеческая клетка» . www.proteinatlas.org . Архивировано из оригинала 2 октября 2017 года . Проверено 2 октября 2017 г.
- ^ Тул П.Дж., Окессон Л., Викинг М., Махдессиан Д., Геладаки А., Айт Блал Х. и др. (май 2017 г.). «Субклеточная карта протеома человека». Наука . 356 (6340): eaal3321. дои : 10.1126/science.aal3321 . ПМИД 28495876 . S2CID 10744558 .
- ^ Ай Ф, Нобл WS (сентябрь 2015 г.). «Методы анализа для изучения 3D-архитектуры генома» . Геномная биология . 16 (1): 183. дои : 10.1186/s13059-015-0745-7 . ПМК 4556012 . ПМИД 26328929 .
- ^ Крыштафович А, Шведе Т, Топф М, Фиделис К, Моулт Дж (2019). «Критическая оценка методов прогнозирования структуры белка (CASP) – XIII раунд» . Белки . 87 (12): 1011–1020. дои : 10.1002/прот.25823 . ПМЦ 6927249 . ПМИД 31589781 .
- ^ «Дом – CASP14» . Precisioncenter.org . Архивировано из оригинала 30 января 2023 года . Проверено 12 июня 2023 г.
- ^ Хой Дж.А., Робинсон Х., Трент Дж.Т., Какар С., Смагге Б.Дж., Харгроув М.С. (август 2007 г.). «Растительные гемоглобины: молекулярная летопись окаменелостей эволюции транспорта кислорода». Журнал молекулярной биологии . 371 (1): 168–79. дои : 10.1016/j.jmb.2007.05.029 . ПМИД 17560601 .
- ^ Джампер Дж., Эванс Р., Притцель А., Грин Т., Фигурнов М., Роннебергер О. и др. (август 2021 г.). «Высокоточное предсказание структуры белка с помощью AlphaFold» . Природа . 596 (7873): 583–589. Бибкод : 2021Natur.596..583J . дои : 10.1038/s41586-021-03819-2 . ISSN 1476-4687 . ПМЦ 8371605 . PMID 34265844 .
- ^ «База данных структуры белков AlphaFold» . Alphafold.ebi.ac.uk . Архивировано из оригинала 24 июля 2021 года . Проверено 10 октября 2022 г.
- ^ Титц Б., Раджагопала С.В., Голл Дж., Хойзер Р., МакКевитт М.Т., Палцкилл Т. и др. (май 2008 г.). Зал N (ред.). «Бинарный белковый интерактом бледной трепонемы — спирохеты сифилиса» . ПЛОС ОДИН . 3 (5): e2292. Бибкод : 2008PLoSO...3.2292T . дои : 10.1371/journal.pone.0002292 . ПМК 2386257 . ПМИД 18509523 .
- ^ Jump up to: Перейти обратно: а б «Фонд открытой биоинформатики: О нас» . Официальный сайт . Открытый фонд биоинформатики . Архивировано из оригинала 12 мая 2011 года . Проверено 10 мая 2011 г.
- ^ «Фонд открытой биоинформатики: BOSC» . Официальный сайт . Открытый фонд биоинформатики . Архивировано из оригинала 18 июля 2011 года . Проверено 10 мая 2011 г.
- ^ Нисбет Р., Элдер И.В. Дж., Майнер Г. (2009 г.). «Биоинформатика» . Справочник по приложениям статистического анализа и интеллектуального анализа данных . Академическая пресса. п. 328. ИСБН 978-0-08-091203-5 .
- ^ Канцелярия комиссара. «Развитие нормативной науки – 24–25 сентября 2014 г. Открытый семинар: стандарты секвенирования следующего поколения» . www.fda.gov . Архивировано из оригинала 14 ноября 2017 года . Проверено 30 ноября 2017 г.
- ^ Симонян В., Гёкс Дж., Мазумдер Р. (2017). «Биокомпьютерные объекты — шаг к оценке и проверке биомедицинских научных вычислений» . PDA Журнал фармацевтической науки и технологий . 71 (2): 136–146. дои : 10.5731/pdajpst.2016.006734 . ПМК 5510742 . ПМИД 27974626 .
- ^ Канцелярия комиссара. «Развитие нормативно-правовой науки – разработка на уровне сообщества стандартов HTS для проверки данных и вычислений и поощрения функциональной совместимости» . www.fda.gov . Архивировано из оригинала 26 января 2018 года . Проверено 30 ноября 2017 г.
- ^ Альтеровиц Г., Дин Д., Гобл С., Крузо М.Р., Сойланд-Рейес С., Белл А. и др. (декабрь 2018 г.). «Возможность прецизионной медицины посредством стандартного обмена информацией о происхождении, анализе и результатах HTS» . ПЛОС Биология . 16 (12): e3000099. дои : 10.1371/journal.pbio.3000099 . ПМК 6338479 . ПМИД 30596645 .
- ^ Проект BioCompute Object (BCO) — это совместная и управляемая сообществом платформа для стандартизации вычислительных данных HTS. 1. Документ спецификации BCO: руководство пользователя для понимания и создания B. , biocompute-objects, 3 сентября 2017 г., заархивировано из оригинала 27 июня 2018 г. , получено 30 ноября 2017 г.
- ^ Кэмпбелл AM (1 июня 2003 г.). «Публичный доступ для преподавания геномики, протеомики и биоинформатики» . Образование в области клеточной биологии . 2 (2): 98–111. дои : 10.1187/cbe.03-02-0007 . ПМК 162192 . ПМИД 12888845 .
- ^ Аренас М (сентябрь 2021 г.). «Общие соображения по практике онлайн-обучения биоинформатике во времена COVID-19» . Образование в области биохимии и молекулярной биологии . 49 (5): 683–684. дои : 10.1002/bmb.21558 . ISSN 1470-8175 . ПМЦ 8426940 . ПМИД 34231941 .
- ^ Баркер Д., Ферье Д.Э., Холланд П.В., Митчелл Дж.Б., Плезье Х., Ричи М.Г. и др. (август 2013 г.). «4273π: образование в области биоинформатики на недорогом оборудовании ARM» . БМК Биоинформатика . 13 :522. дои : 10.1186/1471-2105-14-243 . ПМЦ 3751261 . ПМИД 23937194 .
- ^ Баркер Д., Олдерсон Р.Г., МакДонах Дж.Л., Плезье Х., Комри М.М., Дункан Л. и др. (2015). «Практическая деятельность по биоинформатике на университетском уровне приносит пользу добровольным группам учащихся последних двух лет обучения» . Международный журнал STEM-образования . 2 (17). дои : 10.1186/s40594-015-0030-z . hdl : 10023/7704 . S2CID 256396656 .
- ^ МакДонах Дж.Л., Баркер Д., Олдерсон Р.Г. (2016). «Привлечение вычислительной науки к публике» . СпрингерПлюс . 5 (259): 259. дои : 10.1186/s40064-016-1856-7 . ПМЦ 4775721 . ПМИД 27006868 .
- ^ Робсон Дж. Ф., Баркер Д. (октябрь 2015 г.). «Сравнение содержания белок-кодирующих генов Chlamydia trachomatis и Protochlamydia amoebophila с использованием компьютера Raspberry Pi» . Исследовательские заметки BMC . 8 (561): 561. дои : 10.1186/s13104-015-1476-2 . ПМК 4604092 . ПМИД 26462790 .
- ^ Реггельсворт К.М., Баркер Д. (октябрь 2015 г.). «Сравнение кодирующих белок геномов двух зеленых серобактерий, Chlorobium tepidum TLS и Pelodictyon phaeoclathratiforme BU-1» . Исследовательские заметки BMC . 8 (565): 565. дои : 10.1186/s13104-015-1535-8 . ПМК 4606965 . ПМИД 26467441 .
Дальнейшее чтение [ править ]
- Сегал и др. : Структурные, филогенетические и стыковочные исследования активатора оксидазы D-аминокислот (DAOA), гена-кандидата шизофрении. Теоретическая биология и медицинское моделирование 2013 10:3.
- Ачутсанкар С. Наир Вычислительная биология и биоинформатика - краткий обзор. Архивировано 16 декабря 2008 г. в Wayback Machine , Communications of Computer Society of India, январь 2007 г.
- Алуру, Шринивас , изд. Справочник по вычислительной молекулярной биологии . Чепмен и Холл/Crc, 2006. ISBN 1-58488-406-1 (Серия Chapman & Hall/Crc Computer and Information Science)
- Балди П. и Брунак С. Биоинформатика: подход к машинному обучению , 2-е издание. МИТ Пресс, 2001. ISBN 0-262-02506-X
- Барнс, М.Р. и Грей, И.С., ред., Биоинформатика для генетиков , первое издание. Уайли, 2003. ISBN 0-470-84394-2
- Баксеванис А.Д. и Уэллетт БФФ, ред., Биоинформатика: Практическое руководство по анализу генов и белков , третье издание. Уайли, 2005. ISBN 0-471-47878-4
- Баксеванис А.Д., Пецко Г.А., Штейн Л.Д. и Стормо Г.Д., ред., Современные протоколы в биоинформатике . Уайли, 2007. ISBN 0-471-25093-7
- Кристианини Н. и Хан М. Введение в вычислительную геномику. Архивировано 4 января 2009 г. в Wayback Machine , Cambridge University Press, 2006. ( ISBN 9780521671910 | ISBN 0-521-67191-4 )
- Дурбин Р., С. Эдди, А. Крог и Г. Митчисон, Анализ биологических последовательностей . Издательство Кембриджского университета, 1998. ISBN 0-521-62971-3
- Гилберт Д. (сентябрь 2004 г.). «Программные ресурсы по биоинформатике» . Брифинги по биоинформатике . 5 (3): 300–4. дои : 10.1093/нагрудник/5.3.300 . ПМИД 15383216 .
- Кидуэлл Э. Интеллектуальная биоинформатика: применение методов искусственного интеллекта к проблемам биоинформатики . Уайли, 2005. ISBN 0-470-02175-6
- Кохане и др. Микрочипы для интегративной геномики. Массачусетский технологический институт Пресс, 2002. ISBN 0-262-11271-X
- Лунд О. и др. Иммунологическая биоинформатика. Массачусетский технологический институт Пресс, 2005. ISBN 0-262-12280-4
- Пахтер, Лиор и Штурмфельс, Бернд . «Алгебраическая статистика для вычислительной биологии», издательство Кембриджского университета, 2005. ISBN 0-521-85700-7
- Певзнер, Павел А. Вычислительная молекулярная биология: алгоритмический подход. MIT Press, 2000. ISBN 0-262-16197-4
- Сойнов, Л. Биоинформатика и распознавание образов объединяются. Архивировано 10 мая 2013 г. в журнале Wayback Machine Journal of Pattern Recognition Research ( JPRR. Архивировано 8 сентября 2008 г. в Wayback Machine ), Том 1 (1), 2006 г., стр. 37–41
- Стивенс, Халлам, Жизнь вне последовательности: история биоинформатики, основанная на данных , Чикаго: The University of Chicago Press, 2013, ISBN 9780226080208
- Тисдалл, Джеймс. «Начало Perl для биоинформатики» О'Рейли, 2001. ISBN 0-596-00080-4
- Катализируя исследования на стыке вычислений и биологии (2005 г.), отчет CSTB. Архивировано 28 января 2007 г. в Wayback Machine.
- Вычисление секретов жизни: вклад математических наук и вычислений в молекулярную биологию (1995). Архивировано 6 июля 2008 года в Wayback Machine.
- Курс MIT «Основы вычислительной и системной биологии»
- Вычислительная биология: геномы, сети, эволюция. Бесплатный курс MIT. Архивировано 8 апреля 2013 г. в Wayback Machine.
