Спектроскопия ядерно-магнитного резонанса белков
Спектроскопия ядерного магнитного резонанса белков (обычно сокращенно ЯМР белков ) — область структурной биологии , в которой ЯМР-спектроскопия используется для получения информации о структуре и динамике белков , а также нуклеиновых кислот и их комплексов. Пионерами в этой области были Ричард Р. Эрнст и Курт Вютрих в ETH . [1] и Ад Бакс , Мариус Клор , Анджела Гроненборн из НИЗ , [2] и Герхард Вагнер из Гарвардского университета и другие. Определение структуры методом ЯМР-спектроскопии обычно состоит из нескольких этапов, на каждом из которых используется отдельный набор узкоспециализированных методов. Подготавливается образец, проводятся измерения, применяются интерпретационные подходы, а структура рассчитывается и проверяется.
ЯМР включает квантово-механические свойства центрального ядра (« ядра ») атома. Эти свойства зависят от локального молекулярного окружения, и их измерение дает представление о том, как атомы химически связаны, насколько близко они расположены в пространстве и как быстро они движутся относительно друг друга. Эти свойства в основном такие же, как и в более знакомой магнитно-резонансной томографии (МРТ) , но молекулярные приложения используют несколько иной подход, подходящий для изменения масштаба от миллиметров (интересующих радиологов) до нанометров (связанные атомы обычно разница составляет доли нанометра), в миллион раз. Такое изменение масштаба требует гораздо более высокой чувствительности обнаружения и стабильности для долгосрочных измерений. В отличие от МРТ, исследования структурной биологии не создают изображение напрямую, а полагаются на сложные компьютерные расчеты для создания трехмерных молекулярных моделей.
В настоящее время большинство образцов исследуются в водном растворе, но разрабатываются методы работы и с твердыми образцами . Сбор данных основан на помещении образца в мощный магнит, отправке радиочастотных сигналов через образец и измерении поглощения этих сигналов. В зависимости от окружения атомов внутри белка ядра отдельных атомов будут поглощать радиосигналы разных частот. Кроме того, сигналы поглощения разных ядер могут искажаться соседними ядрами. Эту информацию можно использовать для определения расстояния между ядрами. Эти расстояния, в свою очередь, можно использовать для определения общей структуры белка.
Типичное исследование может включать в себя взаимодействие двух белков друг с другом, возможно, с целью разработки небольших молекул, которые можно будет использовать для исследования нормальной биологии взаимодействия (« химическая биология ») или для выявления возможных направлений для фармацевтического использования ( разработка лекарств) . ). Часто взаимодействующая пара белков может быть идентифицирована в ходе исследований генетики человека, что указывает на то, что взаимодействие может быть нарушено неблагоприятными мутациями, или же они могут играть ключевую роль в нормальной биологии «модельного» организма, такого как плодовая мушка, дрожжи. , червь C. elegans , или мыши. Для подготовки образца обычно используются методы молекулярной биологии, которые производятся путем бактериальной ферментации . Это также позволяет изменять изотопный состав молекулы, что желательно, поскольку изотопы ведут себя по-разному и обеспечивают методы идентификации перекрывающихся сигналов ЯМР.
Подготовка проб
[ редактировать ]
Белковый ядерный магнитный резонанс проводят на водных образцах высокоочищенного белка . Обычно объем пробы составляет от 300 до 600 микролитров с концентрацией белка в диапазоне 0,1–3 миллимолярной . Источник белка может быть как природным, так и произведенным в производственной системе с использованием рекомбинантной ДНК методов посредством генной инженерии . Рекомбинантно экспрессируемые белки обычно легче производить в достаточном количестве, и этот метод делает возможным изотопное мечение . [ нужна ссылка ]
Очищенный белок обычно растворяют в буферном растворе и доводят до желаемых условий растворителя. Образец ЯМР готовят в тонкостенной стеклянной трубке . [ нужна ссылка ]
Сбор данных
[ редактировать ]ЯМР белка использует многомерные эксперименты ядерного магнитного резонанса для получения информации о белке. В идеале каждое отдельное ядро в молекуле находится в отдельном электронном окружении и, следовательно, имеет особый химический сдвиг , по которому его можно распознать. Однако в больших молекулах, таких как белки, число резонансов обычно может достигать нескольких тысяч, и одномерный спектр неизбежно имеет случайные перекрытия. Поэтому проводятся многомерные эксперименты, которые коррелируют частоты различных ядер. Дополнительные измерения уменьшают вероятность перекрытия и имеют большую информативность, поскольку они коррелируют сигналы от ядер внутри определенной части молекулы. Намагниченность передается в образец с помощью импульсов электромагнитной ( радиочастотной ) энергии и между ядрами с помощью задержек; процесс описывается так называемыми импульсными последовательностями . Последовательности импульсов позволяют экспериментатору исследовать и выбирать конкретные типы связей между ядрами. Множество экспериментов по ядерному магнитному резонансу, проводимых на белках, делятся на две основные категории: одна, где намагниченность передается через химические связи, и другая, где передача осуществляется через пространство, независимо от структуры связи. Первая категория используется для присвоения различных химические сдвиги к определенному ядру, а второй в основном используется для создания ограничений расстояния, используемых при расчете структуры и при назначении немеченого белка. [ нужна ссылка ]
В зависимости от концентрации образца, магнитного поля спектрометра и типа эксперимента, одиночный многомерный эксперимент по ядерному магнитному резонансу на образце белка может занять часы или даже несколько дней, чтобы получить подходящее соотношение сигнал/шум через сигнал. усреднение и обеспечение достаточной эволюции переноса намагниченности в различных измерениях эксперимента. При прочих равных условиях эксперименты в более высоких измерениях займут больше времени, чем эксперименты в более низких измерениях. [ нужна ссылка ]
Обычно первый эксперимент, который необходимо измерить с меченным изотопом белком, представляет собой спектр двумерной гетероядерной одноквантовой корреляции (HSQC), где «гетероядерный» относится к ядрам, отличным от 1H. Теоретически гетероядерная одноквантовая корреляция имеет один пик для каждого H, связанного с гетероядером. Так, в 15N-HSQC с 15 N- меченный белок, для каждого атома азота в основной цепи ожидается один сигнал, за исключением пролина , который не имеет амидного водорода из-за циклической природы его основной цепи. Дополнительные сигналы 15N-HSQC вносят каждый остаток с азотно-водородной связью в боковой цепи (W, N, Q, R, H, K). 15N-HSQC часто называют «отпечатком пальца» белка, поскольку каждый белок имеет уникальный набор сигнальных положений. Анализ 15N-HSQC позволяет исследователям оценить, присутствует ли ожидаемое количество пиков, и, таким образом, выявить возможные проблемы, связанные с множественными конформациями или гетерогенностью образца. Относительно быстрый гетероядерный одноквантовый корреляционный эксперимент помогает определить возможность проведения последующих более длительных, более дорогих и более сложных экспериментов. Невозможно приписать пики конкретным атомам только на основе гетероядерной одноквантовой корреляции. [ нужна ссылка ]
Назначение резонанса
[ редактировать ]Для анализа данных ядерного магнитного резонанса важно получить резонансное назначение белка, то есть выяснить, какой химический сдвиг какому атому соответствует. Обычно это достигается путем последовательной ходьбы с использованием информации, полученной в результате нескольких различных типов экспериментов ЯМР. Точная процедура зависит от того, помечен ли белок изотопно или нет, поскольку многие эксперименты по определению зависят от углерода-13 и азота-15. [ нужна ссылка ]
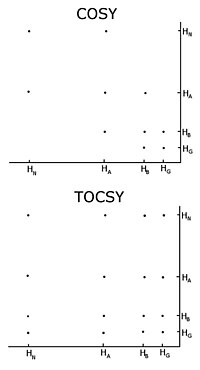
Гомоядерный ядерный магнитный резонанс
[ редактировать ]С немеченым белком обычная процедура состоит в регистрации серии двумерных экспериментов по гомоядерному ядерному магнитному резонансу с помощью корреляционной спектроскопии (COSY), несколько типов которых включают традиционную корреляционную спектроскопию, полную корреляционную спектроскопию (TOCSY) и ядерного Оверхаузера спектроскопию (NOESY). . [3] [4] Двумерный эксперимент по ядерному магнитному резонансу дает двумерный спектр. Единицами обеих осей являются химические сдвиги. COSY и TOCSY передают намагниченность посредством химических связей между соседними протонами. Обычный эксперимент по корреляционной спектроскопии способен передавать намагниченность только между протонами соседних атомов, тогда как в эксперименте по полной корреляционной спектроскопии протоны способны передавать намагниченность, поэтому она передается между всеми протонами, связанными соседними атомами. Таким образом, в традиционной корреляционной спектроскопии альфа-протон передает намагниченность бета-протонам, бета-протоны передаются альфа- и гамма-протонам, если таковые имеются, затем гамма-протон передается бета- и дельта-протонам, и процесс продолжается. . В полной корреляционной спектроскопии альфа и все остальные протоны способны передавать намагниченность бета, гамма, дельта, эпсилон, если они соединены непрерывной цепочкой протонов. Непрерывная цепочка протонов представляет собой боковую цепь отдельного человека. аминокислоты . Таким образом, эти два эксперимента используются для построения так называемых спиновых систем, то есть построения списка резонансов химического сдвига протона пептида, альфа-протонов и всех протонов из боковой цепи каждого остатка . Какие химические сдвиги соответствуют каким ядрам в спиновой системе, определяется традиционными связями корреляционной спектроскопии и тем фактом, что разные типы протонов обладают характерными химическими сдвигами. Чтобы соединить различные спиновые системы в последовательном порядке, необходимо использовать эксперимент по спектроскопии ядерного эффекта Оверхаузера. Поскольку этот эксперимент переносит намагниченность через пространство, он покажет перекрестные пики для всех протонов, находящихся близко в пространстве, независимо от того, находятся ли они в одной спиновой системе или нет. Соседние остатки по своей природе близки в пространстве, поэтому отнесение можно производить по пикам NOESY с другими спиновыми системами. [ нужна ссылка ]
Одной из важных проблем использования гомоядерного ядерного магнитного резонанса является перекрытие пиков. Это происходит, когда разные протоны имеют одинаковые или очень похожие химические сдвиги. Эта проблема усугубляется по мере того, как белок становится больше, поэтому гомоядерный ядерный магнитный резонанс обычно ограничивается небольшими белками или пептидами. [ нужна ссылка ]
Ядерный магнитный резонанс азота-15
[ редактировать ]Наиболее часто выполняемый эксперимент с 15N - это 1 ЧАС- 15 Н HSQC. Эксперимент обладает высокой чувствительностью и поэтому может быть проведен относительно быстро. Его часто используют для проверки пригодности белка для определения структуры с помощью ЯМР, а также для оптимизации условий образца. Это один из стандартных наборов экспериментов, используемых для определения структуры раствора белка. HSQC может быть дополнительно расширен до трех- и четырехмерных экспериментов ЯМР, таких как 15 N-TOCSY-HSQC и 15 Н-НОЭЗИ-HSQC. [5]

Ядерный магнитный резонанс углерода-13 и азота-15
[ редактировать ]Когда белок помечен углеродом-13 и азотом-15, можно записать эксперименты с тройным резонансом , которые передают намагниченность через пептидную связь и, таким образом, соединяют различные спиновые системы через связи. [6] [7] Обычно это делается с помощью некоторых из следующих экспериментов: ГНКО , HN(CA)CO }, ХНК , [8] HN(CO)CA , HNCACB и CBCA(CO)NH . Все шесть экспериментов состоят из 1 ЧАС- 15 Плоскость N (аналогичная спектру HSQC) расширена за счет углерода. В HN(CA)CO , каждая плоскость H N содержит пики карбонильного углерода своего остатка, а также предыдущего в последовательности. HNCO содержит химический сдвиг карбонильного углерода только от предыдущего остатка, но он гораздо более чувствителен, чем HN(CA)CO . Эти эксперименты позволяют каждому 1 ЧАС- 15 Пик N должен быть связан с предыдущим карбонильным углеродом, и затем можно провести последовательное присвоение путем сопоставления сдвигов собственного и предыдущего атомов углерода каждой спиновой системы. HNCA и HN(CO)CA работает аналогично, только с альфа-углеродами (Cα ) , а не с карбонилами, а HNCACB и CBCA(CO)NH содержит как альфа-углерод, так и бета-углерод (Cβ ) . Обычно требуется несколько таких экспериментов, чтобы устранить перекрытие в углеродном измерении. Эта процедура обычно менее двусмысленна, чем метод, основанный на NOESY, поскольку она основана на сквозной передаче облигаций. В методах, основанных на NOESY, будут появляться дополнительные пики, соответствующие атомам, близким в пространстве, но не принадлежащим последовательным остаткам, что запутывает процесс отнесения. После первоначального последовательного резонансного присвоения обычно можно распространить присвоение от Cα и Cβ до остальной части боковой цепи, используя такие эксперименты, как HCCH-TOCSY, который по сути представляет собой эксперимент TOCSY, разрешенный в дополнительном углеродном измерении.
Поколение сдержанности
[ редактировать ]Для проведения расчетов конструкции необходимо создать ряд экспериментально определенных ограничений. Они делятся на разные категории; Наиболее широко используются ограничители расстояния и угловые ограничения.
Ограничения расстояния
[ редактировать ]Перекрестный пик в эксперименте NOESY означает пространственную близость между двумя рассматриваемыми ядрами. Таким образом, каждый пик можно преобразовать в максимальное расстояние между ядрами, обычно от 1,8 до 6 ангстрем . Интенсивность пика NOESY пропорциональна расстоянию в минус 6-й степени, поэтому расстояние определяется в соответствии с интенсивностью пика. Зависимость интенсивности от расстояния не точна, поэтому обычно используется диапазон расстояний.
Очень важно отнести пики NOESY к правильным ядрам на основе химических сдвигов. Если эта задача выполняется вручную, это обычно очень трудоемко, поскольку белки обычно имеют тысячи пиков NOESY. Некоторые компьютерные программы, такие как PASD [9] [10] / XPLOR-NIH , [11] [12] СОЮЗ , [13] БЕЛЫЙ , [14] ВОЗДУХ [15] / ЦНС , [16] и АУДАНА [17] / ПОНДЕРОЗА-C/S [18] на платформе Интегративного ЯМР [19] выполнять эту задачу автоматически на предварительно обработанных вручную списках положений пиков и пиковых объемов в сочетании с расчетом структуры. Прямой доступ к необработанным данным NOESY без громоздкой необходимости итеративно уточнять списки пиков пока предоставляется только PASD. [10] алгоритм реализован в XPLOR-NIH , [11] подход ATNOS/CANDID, реализованный в программном пакете UNIO, [13] и PONDEROSA-C/S и, таким образом, действительно гарантирует объективный и эффективный спектральный анализ NOESY.
Чтобы получить максимально точные определения, очень полезно иметь доступ к экспериментам NOESY по углероду-13 и азоту-15, поскольку они помогают устранить перекрытие в протонном измерении. Это приводит к более быстрому и надежному выполнению заданий и, в свою очередь, к улучшению структуры.
Угловые ограничения
[ редактировать ]В дополнение к ограничениям по расстоянию могут быть созданы ограничения на торсионные углы химических связей, обычно углы пси и фи . Один из подходов заключается в использовании уравнения Карплюса для создания угловых ограничений на основе констант связи . Другой подход использует химические сдвиги для создания угловых ограничений. Оба метода используют тот факт, что геометрия вокруг альфа-углерода влияет на константы взаимодействия и химические сдвиги, поэтому, учитывая константы взаимодействия или химические сдвиги, можно сделать квалифицированное предположение об углах скручивания.
Ограничения ориентации
[ редактировать ]
Молекулы аналита в образце можно частично упорядочить относительно внешнего магнитного поля спектрометра, манипулируя условиями образца. Обычные методы включают добавление к образцу бактериофагов или бицелл или приготовление образца в растянутом полиакриламидном геле . Это создает локальную среду, которая благоприятствует определенной ориентации несферических молекул. Обычно в ЯМР раствора диполярные связи между ядрами усредняются из-за быстрого переворота молекулы. Небольшое перенаселение одной ориентации означает, что остаточную диполярную связь еще предстоит наблюдать. Диполярная связь обычно используется в ЯМР твердого тела и предоставляет информацию об относительной ориентации векторов связей относительно одной глобальной системы отсчета. Обычно ориентацию вектора NH исследуют в эксперименте, подобном HSQC. Первоначально остаточные диполярные связи использовались для уточнения ранее определенных структур, но были также предприняты попытки определения структуры de novo. [20]
Водородно-дейтериевый обмен
[ редактировать ]ЯМР-спектроскопия специфична для ядра. Таким образом, он может различать водород и дейтерий. Протоны амида в белке легко обмениваются с растворителем, и, если растворитель содержит другой изотоп, обычно дейтерий , реакцию можно контролировать с помощью ЯМР-спектроскопии. Насколько быстро происходит обмен данного амида, отражает его доступность для растворителей. Таким образом, скорость обмена амидов может дать информацию о том, какие части белка скрыты, связаны водородными связями и т. д. Обычное применение - сравнение обмена свободной формы и комплекса. Предполагается, что амиды, которые становятся защищенными в комплексе, находятся на границе взаимодействия.
Расчет конструкции
[ редактировать ]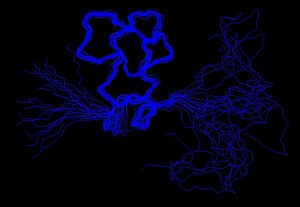
Экспериментально определенные ограничения можно использовать в качестве входных данных для процесса расчета конструкции. Исследователи, используя компьютерные программы, такие как XPLOR-NIH , [11] CYANA , GeNMR или RosettaNMR [21] попытаться удовлетворить как можно больше ограничений в дополнение к общим свойствам белков, таким как длины связей и углы. Алгоритмы преобразуют ограничения и общие свойства белка в энергетические термины, а затем пытаются минимизировать эту энергию. В результате этого процесса образуется ансамбль структур, которые, если данных будет достаточно, чтобы определить определенную складку, сойдутся.
Проверка структуры
[ редактировать ]Полученный ансамбль структур представляет собой «экспериментальную модель», т. е. представление определенного рода экспериментальных данных. Признать этот факт важно, поскольку это означает, что модель может быть хорошим или плохим представлением экспериментальных данных. [22] В целом качество модели будет зависеть как от количества и качества экспериментальных данных, использованных для ее создания, так и от правильной интерпретации таких данных.
Каждый эксперимент имеет связанные с ним ошибки. Случайные ошибки повлияют на воспроизводимость и точность получаемых структур. Если ошибки систематические, это повлияет на точность модели. Прецизионность указывает на степень воспроизводимости измерения и часто выражается как дисперсия набора измеренных данных при одних и тех же условиях. Однако точность указывает на степень, в которой измерение приближается к своему «истинному» значению.
В идеале, модель белка будет тем точнее, чем лучше она будет соответствовать реальной молекуле, которую она представляет, и будет тем точнее, поскольку существует меньшая неопределенность в отношении положения их атомов. На практике не существует «стандартной молекулы», с которой можно было бы сравнивать модели белков, поэтому точность модели определяется степенью согласия между моделью и набором экспериментальных данных. Исторически сложилось так, что структуры, определенные с помощью ЯМР, обычно были более низкого качества, чем структуры, определенные с помощью рентгеновской дифракции. Частично это связано с меньшим объемом информации, содержащейся в данных, полученных методом ЯМР. Из-за этого стало обычной практикой устанавливать качество ансамблей ЯМР путем сравнения его с уникальной конформацией, определенной с помощью дифракции рентгеновских лучей для того же белка. Однако структура дифракции рентгеновских лучей может не существовать, и, поскольку белки в растворе представляют собой гибкие молекулы, белок, представленный одной структурой, может привести к недооценке внутренних изменений атомных положений белка. Набор конформаций, определенный с помощью ЯМР или рентгеновской кристаллографии, может быть лучшим представлением экспериментальных данных белка, чем уникальная конформация. [23]
Полезность модели будет определяться, по крайней мере частично, степенью точности и прецизионности модели. Точная модель с относительно низкой точностью может быть полезна для изучения эволюционных взаимосвязей между структурами набора белков, тогда как рациональный дизайн лекарств требует как точных, так и точных моделей. Неточная модель, независимо от степени точности, с которой она была получена, не будет очень полезной. [22] [24]
Поскольку белковые структуры представляют собой экспериментальные модели, которые могут содержать ошибки, очень важно уметь обнаруживать эти ошибки. Процесс, направленный на обнаружение ошибок, известен как валидация.Существует несколько методов проверки структур: некоторые из них являются статистическими, например PROCHECK и WHAT IF, а другие основаны на физических принципах, таких как CheShift , или на смеси статистических и физических принципов PSVS .
Динамика
[ редактировать ]Помимо структур, ядерный магнитный резонанс может дать информацию о динамике различных частей белка . Обычно это включает измерение времен релаксации, таких как T 1 и T 2, для определения параметров порядка, времени корреляции и скорости химического обмена. Релаксация ЯМР является следствием локальных флуктуаций магнитных полей внутри молекулы. Локальные флуктуирующие магнитные поля генерируются молекулярными движениями. Таким образом, измерения времени релаксации могут предоставить информацию о движениях внутри молекулы на атомном уровне. В ЯМР-исследованиях динамики белков изотоп азота-15 является предпочтительным ядром для изучения, поскольку время его релаксации относительно легко связать с молекулярными движениями. Однако для этого требуется изотопная маркировка белка. Времена релаксации T 1 и T 2 могут быть измерены с использованием различных типов экспериментов на основе HSQC . Типы движений, которые можно обнаружить, — это движения, которые происходят во временном масштабе от примерно 10 пикосекунд до примерно 10 наносекунд. Кроме того, можно изучать и более медленные движения, происходящие в масштабе времени от 10 микросекунд до 100 миллисекунд. Однако, поскольку атомы азота находятся в основном в основной цепи белка, результаты в основном отражают движения основной цепи, которая является наиболее жесткой частью белковой молекулы. Таким образом, результаты, полученные Измерения релаксации азота-15 могут не отражать весь белок. Поэтому недавно были разработаны методы, использующие измерения релаксации углерода-13 и дейтерия , что позволяет систематически изучать движения боковых цепей аминокислот в белках.Сложный и особый случай исследования динамики и гибкости пептидов и полноразмерных белков представляет собой неупорядоченные структуры. В настоящее время общепринятой концепцией является то, что белки могут проявлять более гибкое поведение, известное как беспорядок или отсутствие структуры; однако можно описать ансамбль структур, а не статическую картину, представляющую полностью функциональное состояние белка. В этой области представлены многие достижения, в частности, с точки зрения новых последовательностей импульсов, технологических усовершенствований и тщательной подготовки исследователей в этой области.
ЯМР-спектроскопия крупных белков
[ редактировать ]Традиционно спектроскопия ядерного магнитного резонанса ограничивалась относительно небольшими белками или белковыми доменами. Частично это вызвано проблемами разрешения перекрывающихся пиков в более крупных белках, но эта проблема была решена введением изотопной маркировки и многомерными экспериментами. Другая, более серьезная проблема, заключается в том, что в крупных белках намагниченность релаксирует быстрее, а значит, времени на обнаружение сигнала остается меньше. Это, в свою очередь, приводит к тому, что пики становятся шире и слабее и в конечном итоге исчезают. Для ослабления релаксации были внедрены два метода: спектроскопия, оптимизированная для поперечной релаксации (TROSY). [25] и дейтерирование [26] белков. С помощью этих методов удалось изучить белки в комплексе с шапероном GroES - GroEL массой 900 кДа . [27] [28]
Автоматизация процесса
[ редактировать ]Определение структуры с помощью ЯМР традиционно было трудоемким процессом, требующим интерактивного анализа данных высококвалифицированным ученым. Был значительный интерес к автоматизации процесса, чтобы увеличить производительность определения структуры и сделать ЯМР белков доступным для неспециалистов (см. Структурная геномика ). Двумя наиболее трудоемкими процессами являются назначение резонанса для конкретной последовательности (распределение основной и боковой цепи) и задачи назначения NOE. Было опубликовано несколько различных компьютерных программ, которые автоматизированно нацелены на отдельные части общего процесса определения структуры ЯМР. Наибольший прогресс был достигнут в решении задачи автоматического назначения NOE. До сих пор были предложены только подходы FLYA и UNIO для выполнения всего процесса определения структуры белка ЯМР в автоматическом режиме без какого-либо вмешательства человека. [13] [14] Модули в составе NMRFAM-SPARKY , такие как APES (двухбуквенный код: ae), I-PINE/PINE-SPARKY (двухбуквенный код: ep; веб-сервер I-PINE ) и PONDEROSA (двухбуквенный код: c3, выше; веб-сервер PONDEROSA ) интегрированы таким образом, что обеспечивают полную автоматизацию с возможностью визуальной проверки на каждом этапе. [29] Также были предприняты усилия по стандартизации протокола расчета конструкции, чтобы сделать его более быстрым и поддающимся автоматизации. [30] Недавно был выпущен пакет POKY , преемник упомянутых выше программ, предоставляющий современные инструменты графического пользовательского интерфейса и функции AI/ML. [31]
См. также
[ редактировать ]- ЯМР-спектроскопия
- Ядерный магнитный резонанс
- Спектроскопия ядерного магнитного резонанса углеводов
- Спектроскопия ядерного магнитного резонанса нуклеиновых кислот
- Кристаллизация белка
- Динамика белка
- Релаксация (ЯМР)
- Рентгеновская кристаллография
Ссылки
[ редактировать ]- ^ Вютрих К. (ноябрь 2001 г.). «Путь к ЯМР-структурам белков». Структурная биология природы . 8 (11): 923–925. дои : 10.1038/nsb1101-923 . ПМИД 11685234 . S2CID 26153265 .
- ^ Клор Г.М., Василишен Р.Л., Харрис Р.К. (2011). «Приключения в биомолекулярном ЯМР» (PDF) . В Харрис Р.К., Василишен Р.Л. (ред.). Энциклопедия магнитного резонанса . Джон Уайли и сыновья. дои : 10.1002/9780470034590 . hdl : 11693/53364 . ISBN 9780470034590 .
- ^ Вютрих К. (декабрь 1990 г.). «Определение структуры белка в растворе методом ЯМР-спектроскопии» . Журнал биологической химии . 265 (36): 22059–22062. дои : 10.1016/S0021-9258(18)45665-7 . ПМИД 2266107 .
- ^ Клор Г.М., Гроненборн А.М. (1989). «Определение трехмерной структуры белков и нуклеиновых кислот в растворе методом спектроскопии ядерного магнитного резонанса». Критические обзоры по биохимии и молекулярной биологии . 24 (5): 479–564. дои : 10.3109/10409238909086962 . ПМИД 2676353 .
- ^ Клор ГМ, Гроненборн А.М. (июнь 1991 г.). «Структуры более крупных белков в растворе: трех- и четырехмерная гетероядерная ЯМР-спектроскопия». Наука . 252 (5011): 1390–1399. дои : 10.1126/science.2047852 . ОСТИ 83376 . ПМИД 2047852 .
- ^ Клор Г.М., Гроненборн А.М. (1991). «Применение трех- и четырехмерной гетероядерной ЯМР-спектроскопии для определения структуры белка». Прогресс в спектроскопии ядерного магнитного резонанса . 23 (1): 43–92. дои : 10.1016/0079-6565(91)80002-J .
- ^ Бакс А, Гжесик С (1993). «Методологические достижения в области ЯМР белков». Отчеты о химических исследованиях . 26 (4): 131–138. дои : 10.1021/ar00028a001 .
- ^ Бакс А., Икура М. (май 1991 г.). «Эффективный метод 3D-ЯМР для корреляции резонансов протонов и амидов основной цепи 15N с альфа-углеродом предыдущего остатка в белках, равномерно обогащенных 15N/13C». Журнал биомолекулярного ЯМР . 1 (1): 99–104. дои : 10.1007/BF01874573 . ПМИД 1668719 . S2CID 20037190 .
- ^ Кушевски Дж., Швитерс К.Д., Гарретт Д.С., Берд Р.А., Тьяндра Н., Клор Г.М. (май 2004 г.). «Полностью автоматизированное, высокоустойчивое к ошибкам определение структуры макромолекул на основе многомерных ядерных спектров усиления Оверхаузера и присвоения химических сдвигов». Журнал Американского химического общества . 126 (20): 6258–6273. дои : 10.1021/ja049786h . ПМИД 15149223 .
- ^ Перейти обратно: а б Кушевски Дж. Дж., Тоттунгал Р. А., Клор Г. М., Швитерс К. Д. (август 2008 г.). «Автоматическое устойчивое к ошибкам определение макромолекулярной структуры на основе многомерных ядерных спектров усиления Оверхаузера и присвоения химических сдвигов: повышенная надежность и производительность алгоритма PASD» . Журнал биомолекулярного ЯМР . 41 (4): 221–239. дои : 10.1007/s10858-008-9255-1 . ПМК 2575051 . ПМИД 18668206 .
- ^ Перейти обратно: а б с Швитерс К.Д., Кушевски Дж.Дж., Тяндра Н., Клор Г.М. (январь 2003 г.). «Пакет Xplor-NIH ЯМР для определения молекулярной структуры» . Журнал магнитного резонанса . 160 (1): 65–73. Бибкод : 2003JMagR.160...65S . дои : 10.1016/S1090-7807(02)00014-9 . ПМИД 12565051 .
- ^ Швитерс CD, Кушевски Дж. Дж., Клор Г. М. (2006). «Использование Xplor-NIH для определения молекулярной структуры ЯМР». Прогресс в спектроскопии ядерного магнитного резонанса . 48 (1): 47–62. дои : 10.1016/j.pnmrs.2005.10.001 .
- ^ Перейти обратно: а б с Херрманн Т (2010). «Расчет структуры белка и автоматизированные ограничения NOE». Энциклопедия магнитного резонанса . дои : 10.1002/9780470034590.emrstm1151 . ISBN 978-0470034590 .
- ^ Перейти обратно: а б Гюнтерт П (2004). «Автоматизированный расчет структуры ЯМР с помощью CYANA». Методы ЯМР белков . Методы Мол. Биол. Том. 278. стр. 353–78. CiteSeerX 10.1.1.332.4843 . дои : 10.1385/1-59259-809-9:353 . ISBN 978-1-59259-809-0 . ПМИД 15318003 .
- ^ Рипинг В., Хабек М., Бардьо Б., Бернар А., Маллиавен Т.Е., Нильгес М. (февраль 2007 г.). «ARIA2: автоматизированное присвоение NOE и интеграция данных при расчете структуры ЯМР» . Биоинформатика . 23 (3): 381–382. doi : 10.1093/биоинформатика/btl589 . ПМИД 17121777 .
- ^ Брюнгер А.Т., Адамс П.Д., Клор Г.М., ДеЛано В.Л., Грос П., Гросс-Кунстлеве Р.В. и др. (сентябрь 1998 г.). «Система кристаллографии и ЯМР: новый пакет программного обеспечения для определения структуры макромолекул». Акта Кристаллографика. Раздел D. Биологическая кристаллография . 54 (Часть 5): 905–921. дои : 10.1107/s0907444998003254 . ПМИД 9757107 . S2CID 33910776 .
- ^ Ли В., Пети СМ, Корнилеску Дж., Старк Дж.Л., Маркли Дж.Л. (июнь 2016 г.). «Алгоритм AUDANA для автоматического определения трехмерной структуры белков по данным ЯМР NOE» . Журнал биомолекулярного ЯМР . 65 (2): 51–57. дои : 10.1007/s10858-016-0036-y . ПМЦ 4921114 . ПМИД 27169728 .
- ^ Ли В., Старк Дж.Л., Маркли Дж.Л. (ноябрь 2014 г.). «PONDEROSA-C/S: пакет клиент-серверного программного обеспечения для автоматического определения трехмерной структуры белков» . Журнал биомолекулярного ЯМР . 60 (2–3): 73–75. дои : 10.1007/s10858-014-9855-x . ПМК 4207954 . ПМИД 25190042 .
- ^ Ли В., Корнилеску Г., Дашти Х., Эгбалния Х.Р., Тонелли М., Вестлер В.М. и др. (апрель 2016 г.). «Интегративный ЯМР для биомолекулярных исследований» . Журнал биомолекулярного ЯМР . 64 (4): 307–332. дои : 10.1007/s10858-016-0029-x . ПМЦ 4861749 . ПМИД 27023095 .
- ^ де Альба Э, Тьяндра Н (2004). «Остаточные диполярные связи в определении структуры белка». Методы ЯМР белков . Методы молекулярной биологии. Том. 278. стр. 89–106. дои : 10.1385/1-59259-809-9:089 . ISBN 978-1-59259-809-0 . ПМИД 15317993 .
- ^ Куэнце, Г; Бонно, Р; Леман, Дж. К.; Мейлер, Дж. (5 ноября 2019 г.). «Интегративное моделирование белков в RosettaNMR на основе редких парамагнитных ограничений» . Структура . 27 (11): 1721–1734.e5. doi : 10.1016/j.str.2019.08.012 . ПМЦ 6834914 . ПМИД 31522945 .
- ^ Перейти обратно: а б Ласковский Р.А. (2003). «Структурное обеспечение качества». Структурная биоинформатика . Том. 44. С. 273–303. дои : 10.1002/0471721204.ch14 . ISBN 9780471202004 . ПМИД 12647391 .
{{cite book}}:|journal=игнорируется ( помогите ) - ^ Арнаутова Ю.А., Вила Ю.А., Мартин О.А., Шерага Х.А. (июль 2009 г.). «Что мы можем узнать, вычислив химические сдвиги 13Calpha для рентгеновских моделей белков?» . Акта Кристаллографика. Раздел D. Биологическая кристаллография . 65 (Часть 7): 697–703. дои : 10.1107/S0907444909012086 . ПМК 2703576 . ПМИД 19564690 .
- ^ Спронк К.А., Набуурс С.Б., Кригер Э., Вриенд Г., Вуистер Г.В. (2004). «Подтверждение белковых структур, полученных с помощью ЯМР-спектроскопии». Прогресс в спектроскопии ядерного магнитного резонанса . 45 (3–4): 315–337. дои : 10.1016/j.pnmrs.2004.08.003 .
- ^ Первушин К., Риек Р., Видер Г., Вютрих К. (ноябрь 1997 г.). «Ослабленная релаксация Т2 за счет взаимного подавления диполь-дипольного взаимодействия и анизотропии химического сдвига указывает на путь к ЯМР-структурам очень больших биологических макромолекул в растворе» . Труды Национальной академии наук Соединенных Штатов Америки . 94 (23): 12366–12371. Бибкод : 1997PNAS...9412366P . дои : 10.1073/pnas.94.23.12366 . ПМК 24947 . ПМИД 9356455 .
- ^ Маркус М.А., Дайи К.Т., Мацудайра П., Вагнер Г. (октябрь 1994 г.). «Влияние дейтерирования на скорость релаксации протонов амида в белках. Гетероядерные эксперименты по ЯМР на виллине 14Т». Журнал магнитного резонанса, серия B. 105 (2): 192–195. Бибкод : 1994JMRB..105..192M . дои : 10.1006/jmrb.1994.1122 . ПМИД 7952934 .
- ^ Фио Дж., Бертельсен Э.Б., Хорвич А.Л., Вютрих К. (июль 2002 г.). «ЯМР-анализ комплекса 900К GroEL GroES». Природа . 418 (6894): 207–211. дои : 10.1038/nature00860 . ПМИД 12110894 . S2CID 2451574 .
- ^ Чандак М.С., Накамура Т., Макабе К., Такенака Т., Мукаяма А., Чаудхури Т.К. и др. (июль 2013 г.). «Кинетика H/D-обмена ко-шаперонина GroES Escherichia coli, изученная методами 2D ЯМР и обмена, подавленного ДМСО». Журнал молекулярной биологии . 425 (14): 2541–60. дои : 10.1016/j.jmb.2013.04.008 . ПМИД 23583779 .
- ^ Ли В., Тонелли М., Маркли Дж.Л. (апрель 2015 г.). «NMRFAM-SPARKY: расширенное программное обеспечение для биомолекулярной ЯМР-спектроскопии» . Биоинформатика . 31 (8): 1325–1327. doi : 10.1093/биоинформатика/btu830 . ПМЦ 4393527 . ПМИД 25505092 .
- ^ Лю Г., Шен Ю., Атрея Х.С., Пэриш Д., Шао Ю., Сукумаран Д.К. и др. (июль 2005 г.). «Протокол сбора и анализа данных ЯМР для высокопроизводительного определения структуры белка» . Труды Национальной академии наук Соединенных Штатов Америки . 102 (30): 10487–10492. Бибкод : 2005PNAS..10210487L . дои : 10.1073/pnas.0504338102 . ПМЦ 1180791 . ПМИД 16027363 .
- ^ Ли В., Рахими М., Ли Ю., Чиу А. (март 2021 г.). «POKY: пакет программного обеспечения для многомерного ЯМР и трехмерного расчета структуры биомолекул» . Биоинформатика . 37 (18): 3041–3042. doi : 10.1093/биоинформатика/btab180 . ПМЦ 8479676 . ПМИД 33715003 .
Дальнейшее чтение
[ редактировать ]- Хитченс Т.К., Правило GS (2005). Основы ЯМР-спектроскопии белков (с акцентом на структурную биологию) . Берлин: Шпрингер. ISBN 978-1-4020-3499-2 .
- Тенг Ц (2005). Структурная биология: практические приложения ЯМР . Берлин: Шпрингер. Бибкод : 2005stbi.book.....T . ISBN 978-0-387-24367-2 .
- Рэнс М., Кавана Дж., Фэйрбразер У.Дж., Хант III AW, Скелтон, Нью-Джерси (2007). ЯМР-спектроскопия белков: принципы и практика (2-е изд.). Бостон: Академическая пресса. ISBN 978-0-12-164491-8 .
- Вютрих К (1986). ЯМР белков и нуклеиновых кислот . Нью-Йорк: Уайли. ISBN 978-0-471-82893-8 .
Внешние ссылки
[ редактировать ]- Стратегия на основе NOESY для определения резонансов основной и боковой цепи крупных белков без дейтерирования (протокол)
- Relax Программное обеспечение для анализа динамики ЯМР
- ProSA-web. Архивировано 11 мая 2011 г. в веб-сервисе Wayback Machine для распознавания ошибок в экспериментально или теоретически определенных белковых структурах.
- Определение структуры белка по скудным экспериментальным данным – вводная презентация
- ЯМР белков эксперименты по ЯМР белков