Последовательность сборки
Эта статья нуждается в дополнительных цитатах для проверки . ( октябрь 2017 г. ) |
В биоинформатике сборка последовательностей означает выравнивание и слияние фрагментов более длинной последовательности ДНК с целью восстановления исходной последовательности. [1] Это необходимо, поскольку технология секвенирования ДНК, возможно, не сможет «прочитать» целые геномы за один раз, а скорее считывает небольшие фрагменты длиной от 20 до 30 000 оснований, в зависимости от используемой технологии. [1] Обычно короткие фрагменты (чтения) возникают в результате секвенирования геномной ДНК или транскрипта гена ( EST ). [1]
Проблему последовательной сборки можно сравнить с тем, как взять множество копий книги, пропустить каждую из них через измельчитель с разными резаками и собрать текст книги воедино, просто взглянув на измельченные части. Помимо очевидной сложности этой задачи, есть и дополнительные практические проблемы: в оригинале может быть много повторяющихся абзацев, а некоторые фрагменты могут быть изменены во время уничтожения и содержать опечатки. Также могут быть добавлены отрывки из другой книги, и некоторые обрывки могут оказаться совершенно неузнаваемыми.
Типы
[ редактировать ]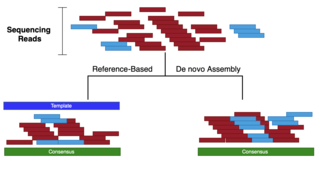
Существует три подхода к сбору данных секвенирования:
- De-novo: сборка считываний секвенирования для создания полноразмерных (иногда новых) последовательностей без использования шаблона (см. « Ассемблеры последовательностей de novo» , «Сборка транскриптома de novo» ). [2]
- Сопоставление/выравнивание: сборка чтений путем выравнивания чтений по шаблону (ссылка AKA). Собранный консенсус может не совпадать с шаблоном.
- На основе ссылки: группировка прочтений по сходству с наиболее похожей областью в пределах ссылки (пошаговое картирование). Затем операции чтения внутри каждой группы сокращаются, чтобы имитировать качество коротких операций чтения. Типичным методом для этого является подход k-mer . Сборка по ссылкам наиболее полезна при использовании длинных чтений . [3]
Сборка по ссылкам представляет собой комбинацию других типов. Этот тип применяется при длинном чтении , чтобы имитировать преимущества короткого считывания (т. е. качество вызова). Логика этого заключается в группировке чтений по меньшим окнам внутри ссылки. Затем чтения в каждой группе будут уменьшены в размере с использованием подхода k-mere для выбора наиболее качественных и наиболее вероятных смежных (контигов). Затем контиги будут соединены вместе, чтобы создать каркас. Окончательное согласие достигается путем закрытия всех щелей в строительных лесах.
Сборки
[ редактировать ]Геном
[ редактировать ]Первые ассемблеры последовательностей начали появляться в конце 1980-х и начале 1990-х годов как варианты более простых программ выравнивания последовательностей , позволяющих собирать воедино огромные количества фрагментов, генерируемых автоматическими инструментами секвенирования, называемыми секвенаторами ДНК . [2] По мере того как секвенированные организмы росли в размерах и усложнялись (от небольших вирусов над плазмидами до бактерий и, наконец, эукариотов ), программы сборки, используемые в этих геномных проектах, требовали все более сложных стратегий для обработки:
- терабайты данных секвенирования, требующие обработки на вычислительных кластерах ;
- идентичные и почти идентичные последовательности (известные как повторы ), которые в худшем случае могут квадратично увеличить временную и пространственную сложность алгоритмов;
- Ошибки чтения ДНК во фрагментах инструментов секвенирования, которые могут затруднить сборку.
Столкнувшись с проблемой сборки первых более крупных геномов эукариот — генома плодовой мухи Drosophila melanogaster в 2000 году и генома человека всего год спустя — учёные разработали такие ассемблеры, как Celera Assembler. [4] и Арахна [5] способен обрабатывать геномы от 130 миллионов (например, плодовой мухи D. melanogaster ) до 3 миллиардов (например, геном человека) пар оснований. После этих усилий несколько других групп, в основном из крупных центров секвенирования генома, создали крупномасштабные ассемблеры и проект с открытым исходным кодом, известный как AMOS. [6] был запущен с целью объединить все инновации в технологии сборки генома в рамках платформы с открытым исходным кодом .

ВОСТОК
[ редактировать ]Метка экспрессируемой последовательности или сборка EST была ранней стратегией, существовавшей с середины 1990-х до середины 2000-х годов, направленной на сборку отдельных генов, а не целых геномов. [7] Эта проблема отличается от сборки генома по нескольким причинам. Входные последовательности для сборки EST представляют собой фрагменты транскрибируемой мРНК клетки и представляют собой лишь часть всего генома. [7] Ряд алгоритмических задач различается между геномом и сборкой EST. Например, геномы часто содержат большое количество повторяющихся последовательностей, сосредоточенных в межгенных областях. Транскрибируемые гены содержат гораздо меньше повторов, что несколько упрощает сборку. С другой стороны, некоторые гены экспрессируются (транскрибируются) в очень больших количествах (например, гены «домашнего хозяйства» ), а это означает, что в отличие от полногеномного секвенирования методом дробовика, считывания не выбираются по всему геному равномерно.
Сборка EST значительно усложняется из-за таких особенностей, как (цис-) альтернативный сплайсинг , транс-сплайсинг , однонуклеотидный полиморфизм и посттранскрипционная модификация . Начиная с 2008 года, когда был изобретен RNA-Seq , секвенирование EST было заменено этой гораздо более эффективной технологией, описанной в разделе « Сборка транскриптома de novo» .
De-novo против сборки сопоставления
[ редактировать ]С точки зрения сложности и требований по времени сборки de novo на несколько порядков медленнее и требуют больше памяти, чем сборки сопоставления. В основном это связано с тем, что алгоритму сборки необходимо сравнивать каждое чтение с каждым другим чтением (операция, имеющая наивную временную сложность O( n 2 )). Современные ассемблеры генома de novo могут использовать различные типы алгоритмов на основе графов, такие как: [8]
- Подход Overlap/Layout/Consensus (OLC), который был типичным для сборщиков данных Сэнгера и основан на графе перекрытия;
- Подход De Bruijn Graph (DBG), который наиболее широко применяется к коротким чтениям с платформ Solexa и SOLiD. Он основан на графах K-mer, которые хорошо работают с огромным количеством коротких чтений;
- Подход на основе жадных графов , который также может использовать один из подходов OLC или DBG. С помощью жадных алгоритмов на основе графов контиги, серии чтений, выровнены вместе. [ нужны дальнейшие объяснения ] , растут за счет жадного расширения, всегда беря на себя чтение, найденное в результате перекрытия с наивысшим баллом. [3]
Ссылаясь на сравнение с измельченными книгами во введении: в то время как для сопоставления сборок в качестве шаблона можно использовать очень похожую книгу (возможно, с измененными именами главных героев и несколькими локациями), сборки de novo представляют собой более сложную задачу. Проблема в том, что нельзя заранее знать, станет ли это научной книгой, романом, каталогом или даже несколькими книгами. Кроме того, каждый клочок будет сравниваться с любым другим клочком.
Обработка повторов при сборке de novo требует построения графа, представляющего соседние повторы. Такую информацию можно получить, прочитав длинный фрагмент, охватывающий повторы полностью или только два его конца . С другой стороны, при сопоставлении сборки детали с несколькими совпадениями или без совпадений обычно оставляются для изучения другим методом сборки. [3]
Технологические достижения
[ редактировать ]Сложность сборки последовательности определяется двумя основными факторами: количеством фрагментов и их длиной. Хотя все больше и более длинные фрагменты позволяют лучше идентифицировать перекрытия последовательностей, они также создают проблемы, поскольку базовые алгоритмы демонстрируют квадратичное или даже экспоненциальное поведение сложности как в отношении количества фрагментов, так и их длины. И хотя более короткие последовательности выравниваются быстрее, они также усложняют этап компоновки сборки, поскольку более короткие чтения сложнее использовать с повторами или почти идентичными повторами.
На заре секвенирования ДНК ученые могли получить лишь несколько последовательностей небольшой длины (несколько десятков оснований) после нескольких недель работы в лабораториях. Следовательно, эти последовательности можно выровнять вручную за несколько минут.
В 1975 году был изобретен метод дидезокси-терминации (он же секвенирование по Сэнгеру ), и вскоре после 2000 года технология была усовершенствована до такой степени, что полностью автоматизированные машины могли производить последовательности в высокопараллельном режиме 24 часа в сутки. Крупные геномные центры по всему миру располагали полными фермами этих машин для секвенирования, что, в свою очередь, привело к необходимости оптимизировать ассемблеры для последовательностей из проектов полногеномного секвенирования , где считываются
- имеют длину около 800–900 оснований
- секвенирования и клонирования содержать артефакты секвенирования, такие как векторы
- имеют уровень ошибок от 0,5 до 10%
Благодаря технологии Сэнгера бактериальные проекты с числом прочтений от 20 000 до 200 000 можно было легко собрать на одном компьютере. Более крупные проекты, такие как изучение генома человека примерно с 35 миллионами чтений, требовали больших вычислительных ферм и распределенных вычислений.
К 2004/2005 году пиросеквенирование довела компания 454 Life Sciences до коммерческой жизнеспособности . [9] Этот новый метод секвенирования генерирует считывания намного короче, чем у секвенирования Сэнгера: первоначально около 100 оснований, теперь 400-500 оснований. [9] Гораздо более высокая производительность и более низкая стоимость (по сравнению с секвенированием по Сэнгеру) подтолкнули к принятию этой технологии центрами генома, что, в свою очередь, подтолкнуло к разработке сборщиков последовательностей, которые могли бы эффективно обрабатывать наборы считываний. Огромный объем данных в сочетании с характерными для технологии шаблонами ошибок при чтении задерживал развитие ассемблеров; в начале 2004 года был доступен только ассемблер Newbler из 454. Выпущенная в середине 2007 года гибридная версия ассемблера MIRA компанией Chevreux et al. [10] был первым свободно доступным ассемблером, который мог собирать 454 чтения, а также смеси 454 чтения и чтения по Сэнгеру. Сборка последовательностей, полученных с помощью различных технологий секвенирования, впоследствии была названа гибридной сборкой . [10]
С 2006 года Illumina доступна технология (ранее Solexa), которая может генерировать около 100 миллионов считываний за один цикл на одном секвенаторе. Сравните это с 35 миллионами чтений проекта генома человека, на создание которого потребовалось несколько лет на сотнях секвенирующих машин. [11] Первоначально Illumina была ограничена длиной всего в 36 оснований, что делало ее менее подходящей для сборки de novo (например, сборка транскриптома de novo ), но новые версии технологии достигают длины чтения более 100 оснований с обоих концов клона длиной 3–400 п.н. . [11] Анонсированный в конце 2007 года ассемблер SHARCGS. [12] Домом и др. был первым опубликованным ассемблером, который использовался для сборки с чтением Solexa. За ним быстро последовал ряд других.
Позже новые технологии, такие как SOLiD от Applied Biosystems , Ion Torrent и SMRT были выпущены новые технологии (например, секвенирование нанопор , и продолжают появляться ). Несмотря на более высокий уровень ошибок в этих технологиях, они важны для сборки, поскольку их большая длина считывания помогает решить проблему повторения. [11] Невозможно собрать идеальный повтор, длина которого превышает максимальную длину чтения; однако по мере того, как чтения становятся длиннее, вероятность такого большого идеального повторения становится малой. Это дает преимущество более длинным прочтениям секвенирования при сборке повторов, даже если они имеют низкую точность (~ 85%). [11]
Контроль качества
[ редактировать ]Большинство ассемблеров последовательностей имеют встроенные алгоритмы контроля качества, например Phred . [13] Однако такие меры не позволяют оценить полноту сборки с точки зрения содержания генов. Некоторые инструменты оценивают качество сборки постфактум.
Например, BUSCO (Бенчмаркинг универсальных однокопийных ортологов) — это мера полноты генов в геноме, наборе генов или транскриптоме , использующая тот факт, что многие гены присутствуют только в виде генов с одной копией в большинстве геномов. [14] Первоначальные наборы BUSCO представляли собой 3023 гена позвоночных , 2675 генов членистоногих , 843 генов многоклеточных животных , 1438 генов грибов и 429 генов эукариот . В этой таблице показан пример геномов человека и плодовой мухи: [14]
| Разновидность | гены | С: | Д: | Ф: | М: | н: |
|---|---|---|---|---|---|---|
| Мудрый человек | 20,364 | 99 | 1.7 | 0.0 | 0.0 | 3,023 |
| Дрозофила меланогастер | 13,918 | 99 | 3.7 | 0.2 | 0.0 | 2,675 |
Алгоритмы сборки
[ редактировать ]Различные организмы имеют в своем геноме отдельные области более высокой сложности. Следовательно, необходимы различные вычислительные подходы. Некоторые из часто используемых алгоритмов:
- Сборка графов: основана на теории графов в информатике. Граф де Брёйна является примером этого подхода и использует k-меры для сборки смежных чтений. [15]
- Сборка жадного графа: этот подход оценивает каждое добавленное чтение к сборке и выбирает максимально возможный балл из перекрывающейся области.
Учитывая набор фрагментов последовательности, цель состоит в том, чтобы найти более длинную последовательность, содержащую все фрагменты (см. рисунок в разделе « Типы сборки последовательностей» ):
- Вычислить попарные выравнивания всех фрагментов.
- Выберите два фрагмента с наибольшим перекрытием.
- Объедините выбранные фрагменты.
- Повторяйте шаги 2 и 3, пока не останется только один фрагмент.
Результат может оказаться не оптимальным решением проблемы.
Конвейер биоинформатики
[ редактировать ]В общем, сборка считываний секвенирования в каркас состоит из трех этапов:
- Предварительная сборка: этот шаг необходим для обеспечения целостности последующего анализа, такого как вызов вариантов или окончательная последовательность каркаса. Этот шаг состоит из двух хронологических рабочих процессов:
- Проверка качества: В зависимости от типа технологии секвенирования могут возникнуть различные ошибки, которые могут привести к ложному основанию . Например, секвенирование «НААААААААААААН» и «НААААААААААН», которые включают 12 аденинов, может быть ошибочно названо вместо них 11 аденином. Секвенирование высокоповторяющегося сегмента целевой ДНК/РНК может привести к тому, что вызов будет на одно основание короче или на одно основание длиннее. Качество чтения обычно измеряется Phred , который представляет собой закодированную оценку качества каждого нуклеотида в последовательности чтения. Некоторые технологии секвенирования, такие как PacBio, не имеют метода оценки секвенированных считываний. На этом этапе обычно используется инструмент FastQC. [16]
- Фильтрация операций чтения: операции чтения, не прошедшие проверку качества, следует удалить из файла FASTQ , чтобы получить наилучшие конфиги сборки.
- Сборка: на этом этапе выравнивание считываний будет использоваться с различными критериями для сопоставления каждого считывания с возможным местоположением. Прогнозируемая позиция чтения основана либо на том, насколько его последовательность совпадает с другими чтениями, либо на ссылке. Для чтения из разных технологий секвенирования используются разные алгоритмы выравнивания. Некоторые из часто используемых подходов при сборке — это граф де Брёйна и перекрытие. Длина чтения, охват , качество и используемый метод секвенирования играют важную роль в выборе лучшего алгоритма выравнивания в случае секвенирования следующего поколения . [17] С другой стороны, алгоритмы, выравнивающие считывания секвенирования третьего поколения, требуют предварительных подходов для учета связанной с ними высокой частоты ошибок.
- Пост-сборка: на этом этапе основное внимание уделяется извлечению ценной информации из собранной последовательности. Сравнительная геномика и популяционный анализ являются примерами анализа после сборки.
Программы
[ редактировать ]Списки ассемблеров de novo см. в разделе « Ассемблеры последовательностей de novo» . Список выравнивателей картирования см. в разделе «Список программного обеспечения для выравнивания последовательностей § Выравнивание последовательностей короткого считывания» .
Некоторые из распространенных инструментов, используемых на различных этапах сборки, перечислены в следующей таблице:
| Программное обеспечение | Тип чтения | Веб-страница инструмента | Примечания |
|---|---|---|---|
| FastQC | Несколько | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | Это распространенный инструмент, используемый для проверки качества считывания различных технологий секвенирования, таких как Illumina , 454 и PacBio . |
| Миссис | Краткое и длинное чтение | https://sourceforge.net/projects/bio-bwa/files/ | Это инструмент командной строки . В основном известен благодаря легкому ходу и точному выравниванию последовательности. |
| МиниКарта2 | Долго читает | https://github.com/lh3/minimap2 | Этот инструмент командной строки предназначен для работы с PacBio и Oxford Nanopore и считывает с вероятностью ошибок 15%. |
| ЛоРеТТА | Долго читает | https://github.com/salvocamiolo/LoReTTA/releases/tag/v0.1 | Этот инструмент предназначен для более точной сборки (на основе справочных материалов) вирусных геномов с использованием считываний PacBio CCS. |
| ЛОПАТЫ | Краткое и длинное чтение | http://cab.spbu.ru/software/spades/ | Это инструмент сборки, который запускается в командной строке. |
| Самтулс | Анализ выравнивания | https://samtools.github.io | Это полезно после сборки. Он может генерировать различную статистику и выполнять несколько этапов фильтрации файла выравнивания. |
См. также
[ редактировать ]- Опять ассемблеры последовательностей
- Выравнивание последовательности
- Сборка транскриптома de novo
- Установить проблему с обложкой
- Список секвенированных геномов животных
- Сборка генома растения
Ссылки
[ редактировать ]- ^ Jump up to: а б с Сон Джи, Нам Дж.В. (январь 2018 г.). «Настоящее и будущее сборки цельного генома de novo». Брифинги по биоинформатике . 19 (1): 23–40. дои : 10.1093/нагрудник/bbw096 . ПМИД 27742661 .
- ^ Jump up to: а б Бейкер М. (27 марта 2012 г.). «Сборка генома de novo: что должен знать каждый биолог» . Природные методы . 9 (4): 333–337. дои : 10.1038/nmeth.1935 . ISSN 1548-7105 .
- ^ Jump up to: а б с Вольф Б. «Сборка генома de novo по сравнению с сопоставлением эталонного генома» (PDF) . Университет прикладных наук Западной Швейцарии . Проверено 6 апреля 2019 г.
- ^ Майерс Э.В., Саттон Г.Г., Делчер А.Л., Дью И.М., Фасуло Д.П., Фланиган М.Дж. и др. (март 2000 г.). «Полногеномная сборка дрозофилы». Наука . 287 (5461): 2196–2204. Бибкод : 2000Sci...287.2196M . CiteSeerX 10.1.1.79.9822 . дои : 10.1126/science.287.5461.2196 . ПМИД 10731133 . S2CID 6049420 .
- ^ Бацоглу С., Джаффе Д.Б., Стэнли К., Батлер Дж., Гнерр С., Маусели Э. и др. (январь 2002 г.). «АРАХНА: сборщик полногеномных дробовиков» . Геномные исследования . 12 (1): 177–189. дои : 10.1101/гр.208902 . ПМК 155255 . ПМИД 11779843 .
- ^ «АМОС ВИКИ» . amos.sourceforge.net . Проверено 2 января 2023 г.
- ^ Jump up to: а б Нагарадж С.Х., Гассер Р.Б., Ранганатан С. (январь 2007 г.). «Руководство для автостопщика по анализу меток выраженной последовательности (EST)». Брифинги по биоинформатике . 8 (1): 6–21. дои : 10.1093/bib/bbl015 . ПМИД 16772268 .
- ^ Ли З, Чен Ю, Му Д, Юань Дж, Ши Ю, Чжан Х и др. (январь 2012 г.). «Сравнение двух основных классов алгоритмов сборки: консенсуса по компоновке перекрытия и графа де-Брюйна». Брифинги по функциональной геномике . 11 (1): 25–37. дои : 10.1093/bfgp/elr035 . ПМИД 22184334 .
- ^ Jump up to: а б Харрингтон Коннектикут, Лин Э.И., Олсон М.Т., Эшлеман-младший (сентябрь 2013 г.). «Основы пиросеквенирования». Архивы патологии и лабораторной медицины . 137 (9): 1296–1303. дои : 10.5858/arpa.2012-0463-RA . ПМИД 23991743 .
- ^ Jump up to: а б "МИРА 2.9.8 для 454 и 454/Sanger гибридной сборки" . groups.google.com . Проверено 2 января 2023 г.
- ^ Jump up to: а б с д Ху Т., Читнис Н., Монос Д., Динь А. (ноябрь 2021 г.). «Технологии секвенирования нового поколения: обзор». Иммунология человека . Секвенирование нового поколения и его применение в медицинской лабораторной иммунологии. 82 (11): 801–811. дои : 10.1016/j.humimm.2021.02.012 . PMID 33745759 .
- ^ Дом Дж.К., Лоттаз С., Бородина Т., Химмельбауэр Х. (ноябрь 2007 г.). «SHARCGS, быстрый и высокоточный алгоритм короткого чтения для геномного секвенирования de novo» . Геномные исследования . 17 (11): 1697–1706. дои : 10.1101/гр.6435207 . ПМК 2045152 . ПМИД 17908823 .
- ^ Кок Пи Джей, Филдс Си Джей, Гото Н, Хойер МЛ, Райс ПМ (апрель 2010 г.). «Формат файла Sanger FASTQ для последовательностей с показателями качества и варианты Solexa/Illumina FASTQ» . Исследования нуклеиновых кислот . 38 (6): 1767–1771. дои : 10.1093/нар/gkp1137 . ПМЦ 2847217 . ПМИД 20015970 .
- ^ Jump up to: а б Симау Ф.А., Уотерхаус Р.М., Иоаннидис П., Кривенцева Е.В., Здобнов Е.М. (октябрь 2015 г.). «BUSCO: оценка сборки генома и полноты аннотаций с помощью однокопийных ортологов». Биоинформатика . 31 (19): 3210–3212. doi : 10.1093/биоинформатика/btv351 . ПМИД 26059717 .
- ^ Компо П.Е., Певзнер П.А., Теслер Г. (ноябрь 2011 г.). «Как применять графы де Брейна для сборки генома» . Природная биотехнология . 29 (11): 987–991. дои : 10.1038/nbt.2023 . ПМЦ 5531759 . ПМИД 22068540 .
- ^ «Биоинформатика Babraham - FastQC Инструмент контроля качества для данных последовательностей с высокой пропускной способностью» . www.bioinformatics.babraham.ac.uk . Проверено 9 мая 2022 г.
- ^ Руффало М., ЛаФрамбуаз Т., Коютюрк М. (октябрь 2011 г.). «Сравнительный анализ алгоритмов выравнивания чтения секвенирования нового поколения» . Биоинформатика . 27 (20): 2790–2796. doi : 10.1093/биоинформатика/btr477 . ПМИД 21856737 .