РНК-Seq

RNA-Seq (названный как сокращение от секвенирования РНК ) — это метод, который использует секвенирование следующего поколения для выявления присутствия и количества молекул РНК в биологическом образце, обеспечивая моментальный снимок экспрессии генов в образце, также известный как транскриптом . [2] [3]
В частности, RNA-Seq облегчает возможность изучения альтернативных транскриптов сплайсинга генов , посттранскрипционных модификаций , слияния генов , мутаций/ SNP и изменений в экспрессии генов с течением времени или различий в экспрессии генов в разных группах или методах лечения. [4] Помимо транскриптов мРНК, RNA-Seq может исследовать различные популяции РНК, включая общую РНК, малые РНК, такие как микроРНК , тРНК и профилирование рибосом . [5] RNA-Seq can also be used to determine exon/intron boundaries and verify or amend previously annotated 5' and 3' gene boundaries. Recent advances in RNA-Seq include single cell sequencing, bulk RNA sequencing,[6] 3' mRNA-sequencing, in situ sequencing of fixed tissue, and native RNA molecule sequencing with single-molecule real-time sequencing.[7] Other examples of emerging RNA-Seq applications due to the advancement of bioinformatics algorithms are copy number alteration, microbial contamination, transposable elements, cell type (deconvolution) and the presence of neoantigens.[8]
Prior to RNA-Seq, gene expression studies were done with hybridization-based microarrays. Issues with microarrays include cross-hybridization artifacts, poor quantification of lowly and highly expressed genes, and needing to know the sequence a priori.[9] Because of these technical issues, transcriptomics transitioned to sequencing-based methods. These progressed from Sanger sequencing of Expressed sequence tag libraries, to chemical tag-based methods (e.g., serial analysis of gene expression), and finally to the current technology, next-gen sequencing of complementary DNA (cDNA), notably RNA-Seq.

Methods
[edit]Library preparation
[edit]
The general steps to prepare a complementary DNA (cDNA) library for sequencing are described below, but often vary between platforms.[10][3][11]
- RNA Isolation: RNA is isolated from tissue and mixed with Deoxyribonuclease (DNase). DNase reduces the amount of genomic DNA. The amount of RNA degradation is checked with gel and capillary electrophoresis and is used to assign an RNA integrity number to the sample. This RNA quality and the total amount of starting RNA are taken into consideration during the subsequent library preparation, sequencing, and analysis steps.
- RNA selection/depletion: To analyze signals of interest, the isolated RNA can either be kept as is, enriched for RNA with 3' polyadenylated (poly(A)) tails to include only eukaryotic mRNA, depleted of ribosomal RNA (rRNA), and/or filtered for RNA that binds specific sequences (RNA selection and depletion methods table, below). RNA molecules having 3' poly(A) tails in eukaryotes are mainly composed of mature, processed, coding sequences. Poly(A) selection is performed by mixing RNA with poly(T) oligomers covalently attached to a substrate, typically magnetic beads.[12][13] Poly(A) selection has important limitations in RNA biotype detection. Many RNA biotypes are not polyadenylated, including many noncoding RNA and histone-core protein transcripts, or are regulated via their poly(A) tail length (e.g., cytokines) and thus might not be detected after poly(A) selection.[14] Furthermore, poly(A) selection may display increased 3' bias, especially with lower quality RNA.[15][16] These limitations can be avoided with ribosomal depletion, removing rRNA that typically represents over 90% of the RNA in a cell. Both poly(A) enrichment and ribosomal depletion steps are labor intensive and could introduce biases, so more simple approaches have been developed to omit these steps.[17] Small RNA targets, such as miRNA, can be further isolated through size selection with exclusion gels, magnetic beads, or commercial kits.
- cDNA synthesis: RNA is reverse transcribed to cDNA because DNA is more stable and to allow for amplification (which uses DNA polymerases) and leverage more mature DNA sequencing technology. Amplification subsequent to reverse transcription results in loss of strandedness, which can be avoided with chemical labeling or single molecule sequencing. Fragmentation and size selection are performed to purify sequences that are the appropriate length for the sequencing machine. The RNA, cDNA, or both are fragmented with enzymes, sonication, divalent ions, or nebulizers. Fragmentation of the RNA reduces 5' bias of randomly primed-reverse transcription and the influence of primer binding sites,[13] with the downside that the 5' and 3' ends are converted to DNA less efficiently. Fragmentation is followed by size selection, where either small sequences are removed or a tight range of sequence lengths are selected. Because small RNAs like miRNAs are lost, these are analyzed independently. The cDNA for each experiment can be indexed with a hexamer or octamer barcode, so that these experiments can be pooled into a single lane for multiplexed sequencing.
| Strategy | Predominant type of RNA | Ribosomal RNA content | Unprocessed RNA content | Isolation method |
|---|---|---|---|---|
| Total RNA | All | High | High | None |
| PolyA selection | Coding | Low | Low | Hybridization with poly(dT) oligomers |
| rRNA depletion | Coding, noncoding | Low | High | Removal of oligomers complementary to rRNA |
| RNA capture | Targeted | Low | Moderate | Hybridization with probes complementary to desired transcripts |
Complementary DNA sequencing (cDNA-Seq)
[edit]The cDNA library derived from RNA biotypes is then sequenced into a computer-readable format. There are many high-throughput sequencing technologies for cDNA sequencing including platforms developed by Illumina, Thermo Fisher, BGI/MGI, PacBio, and Oxford Nanopore Technologies.[18] For Illumina short-read sequencing, a common technology for cDNA sequencing, adapters are ligated to the cDNA, DNA is attached to a flow cell, clusters are generated through cycles of bridge amplification and denaturing, and sequence-by-synthesis is performed in cycles of complementary strand synthesis and laser excitation of bases with reversible terminators. Sequencing platform choice and parameters are guided by experimental design and cost. Common experimental design considerations include deciding on the sequencing length, sequencing depth, use of single versus paired-end sequencing, number of replicates, multiplexing, randomization, and spike-ins.[19]
Small RNA/non-coding RNA sequencing
[edit]When sequencing RNA other than mRNA, the library preparation is modified. The cellular RNA is selected based on the desired size range. For small RNA targets, such as miRNA, the RNA is isolated through size selection. This can be performed with a size exclusion gel, through size selection magnetic beads, or with a commercially developed kit. Once isolated, linkers are added to the 3' and 5' end then purified. The final step is cDNA generation through reverse transcription.
Direct RNA sequencing
[edit]
Because converting RNA into cDNA, ligation, amplification, and other sample manipulations have been shown to introduce biases and artifacts that may interfere with both the proper characterization and quantification of transcripts,[20] single molecule direct RNA sequencing has been explored by companies including Helicos (bankrupt), Oxford Nanopore Technologies,[21] and others. This technology sequences RNA molecules directly in a massively-parallel manner.
Single-molecule real-time RNA sequencing
[edit]Massively parallel single molecule direct RNA-Seq has been explored as an alternative to traditional RNA-Seq, in which RNA-to-cDNA conversion, ligation, amplification, and other sample manipulation steps may introduce biases and artifacts.[22] Technology platforms that perform single-molecule real-time RNA-Seq include Oxford Nanopore Technologies (ONT) Nanopore sequencing,[21] PacBio IsoSeq, and Helicos (bankrupt). Sequencing RNA in its native form preserves modifications like methylation, allowing them to be investigated directly and simultaneously.[21] Another benefit of single-molecule RNA-Seq is that transcripts can be covered in full length, allowing for higher confidence isoform detection and quantification compared to short-read sequencing. Traditionally, single-molecule RNA-Seq methods have higher error rates compared to short-read sequencing, but newer methods like ONT direct RNA-Seq limit errors by avoiding fragmentation and cDNA conversion. Recent uses of ONT direct RNA-Seq for differential expression in human cell populations have demonstrated that this technology can overcome many limitations of short and long cDNA sequencing.[23]
Single-cell RNA sequencing (scRNA-Seq)
[edit]Standard methods such as microarrays and standard bulk RNA-Seq analysis analyze the expression of RNAs from large populations of cells. In mixed cell populations, these measurements may obscure critical differences between individual cells within these populations.[24][25]
Single-cell RNA sequencing (scRNA-Seq) provides the expression profiles of individual cells. Although it is not possible to obtain complete information on every RNA expressed by each cell, due to the small amount of material available, patterns of gene expression can be identified through gene clustering analyses. This can uncover the existence of rare cell types within a cell population that may never have been seen before. For example, rare specialized cells in the lung called pulmonary ionocytes that express the Cystic fibrosis transmembrane conductance regulator were identified in 2018 by two groups performing scRNA-Seq on lung airway epithelia.[26][27]
Experimental procedures
[edit]
Current scRNA-Seq protocols involve the following steps: isolation of single cell and RNA, reverse transcription (RT), amplification, library generation and sequencing. Single cells are either mechanically separated into microwells (e.g., BD Rhapsody, Takara ICELL8, Vycap Puncher Platform, or CellMicrosystems CellRaft) or encapsulated in droplets (e.g., 10x Genomics Chromium, Illumina Bio-Rad ddSEQ, 1CellBio InDrop, Dolomite Bio Nadia).[28] Single cells are labeled by adding beads with barcoded oligonucleotides; both cells and beads are supplied in limited amounts such that co-occupancy with multiple cells and beads is a very rare event. Once reverse transcription is complete, the cDNAs from many cells can be mixed together for sequencing; transcripts from a particular cell are identified by each cell's unique barcode.[29][30] Unique molecular identifier (UMIs) can be attached to mRNA/cDNA target sequences to help identify artifacts during library preparation.[31]
Challenges for scRNA-Seq include preserving the initial relative abundance of mRNA in a cell and identifying rare transcripts.[32] The reverse transcription step is critical as the efficiency of the RT reaction determines how much of the cell's RNA population will be eventually analyzed by the sequencer. The processivity of reverse transcriptases and the priming strategies used may affect full-length cDNA production and the generation of libraries biased toward the 3’ or 5' end of genes.
In the amplification step, either PCR or in vitro transcription (IVT) is currently used to amplify cDNA. One of the advantages of PCR-based methods is the ability to generate full-length cDNA. However, different PCR efficiency on particular sequences (for instance, GC content and snapback structure) may also be exponentially amplified, producing libraries with uneven coverage. On the other hand, while libraries generated by IVT can avoid PCR-induced sequence bias, specific sequences may be transcribed inefficiently, thus causing sequence drop-out or generating incomplete sequences.[33][24]Several scRNA-Seq protocols have been published:Tang et al.,[34]STRT,[35]SMART-seq,[36]CEL-seq,[37]RAGE-seq,[38] Quartz-seq[39] and C1-CAGE.[40] These protocols differ in terms of strategies for reverse transcription, cDNA synthesis and amplification, and the possibility to accommodate sequence-specific barcodes (i.e. UMIs) or the ability to process pooled samples.[41]
In 2017, two approaches were introduced to simultaneously measure single-cell mRNA and protein expression through oligonucleotide-labeled antibodies known as REAP-seq,[42] and CITE-seq.[43]
Applications
[edit]scRNA-Seq is becoming widely used across biological disciplines including Development, Neurology,[44] Oncology,[45][46][47] Autoimmune disease,[48] and Infectious disease.[49]
scRNA-Seq has provided considerable insight into the development of embryos and organisms, including the worm Caenorhabditis elegans,[50] and the regenerative planarian Schmidtea mediterranea.[51][52] The first vertebrate animals to be mapped in this way were Zebrafish[53][54] and Xenopus laevis.[55] In each case multiple stages of the embryo were studied, allowing the entire process of development to be mapped on a cell-by-cell basis.[10] Science recognized these advances as the 2018 Breakthrough of the Year.[56]
Experimental considerations
[edit]A variety of parameters are considered when designing and conducting RNA-Seq experiments:
- Tissue specificity: Gene expression varies within and between tissues, and RNA-Seq measures this mix of cell types. This may make it difficult to isolate the biological mechanism of interest. Single cell sequencing can be used to study each cell individually, mitigating this issue.
- Time dependence: Gene expression changes over time, and RNA-Seq only takes a snapshot. Time course experiments can be performed to observe changes in the transcriptome.
- Coverage (also known as depth): RNA harbors the same mutations observed in DNA, and detection requires deeper coverage. With high enough coverage, RNA-Seq can be used to estimate the expression of each allele. This may provide insight into phenomena such as imprinting or cis-regulatory effects. The depth of sequencing required for specific applications can be extrapolated from a pilot experiment.[57]
- Data generation artifacts (also known as technical variance): The reagents (e.g., library preparation kit), personnel involved, and type of sequencer (e.g., Illumina, Pacific Biosciences) can result in technical artifacts that might be mis-interpreted as meaningful results. As with any scientific experiment, it is prudent to conduct RNA-Seq in a well controlled setting. If this is not possible or the study is a meta-analysis, another solution is to detect technical artifacts by inferring latent variables (typically principal component analysis or factor analysis) and subsequently correcting for these variables.[58]
- Data management: A single RNA-Seq experiment in humans is usually 1-5 Gb (compressed), or more when including intermediate files.[59] This large volume of data can pose storage issues. One solution is compressing the data using multi-purpose computational schemas (e.g., gzip) or genomics-specific schemas. The latter can be based on reference sequences or de novo. Another solution is to perform microarray experiments, which may be sufficient for hypothesis-driven work or replication studies (as opposed to exploratory research).
Analysis
[edit]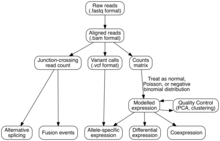
Transcriptome assembly
[edit]Two methods are used to assign raw sequence reads to genomic features (i.e., assemble the transcriptome):
- De novo: This approach does not require a reference genome to reconstruct the transcriptome, and is typically used if the genome is unknown, incomplete, or substantially altered compared to the reference.[60] Challenges when using short reads for de novo assembly include 1) determining which reads should be joined together into contiguous sequences (contigs), 2) robustness to sequencing errors and other artifacts, and 3) computational efficiency. The primary algorithm used for de novo assembly transitioned from overlap graphs, which identify all pair-wise overlaps between reads, to de Bruijn graphs, which break reads into sequences of length k and collapse all k-mers into a hash table.[61] Overlap graphs were used with Sanger sequencing, but do not scale well to the millions of reads generated with RNA-Seq. Examples of assemblers that use de Bruijn graphs are Trinity,[60] Oases[62] (derived from the genome assembler Velvet[63]), Bridger,[64] and rnaSPAdes.[65] Paired-end and long-read sequencing of the same sample can mitigate the deficits in short read sequencing by serving as a template or skeleton. Metrics to assess the quality of a de novo assembly include median contig length, number of contigs and N50.[66]
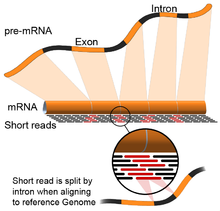
- Genome guided: This approach relies on the same methods used for DNA alignment, with the additional complexity of aligning reads that cover non-continuous portions of the reference genome.[67] These non-continuous reads are the result of sequencing spliced transcripts (see figure). Typically, alignment algorithms have two steps: 1) align short portions of the read (i.e., seed the genome), and 2) use dynamic programming to find an optimal alignment, sometimes in combination with known annotations. Software tools that use genome-guided alignment include Bowtie,[68] TopHat (which builds on BowTie results to align splice junctions),[69][70] Subread,[71] STAR,[67] HISAT2,[72] and GMAP.[73] The output of genome guided alignment (mapping) tools can be further used by tools such as Cufflinks[70] or StringTie[74] to reconstruct contiguous transcript sequences (i.e., a FASTA file). The quality of a genome guided assembly can be measured with both 1) de novo assembly metrics (e.g., N50) and 2) comparisons to known transcript, splice junction, genome, and protein sequences using precision, recall, or their combination (e.g., F1 score).[66] In addition, in silico assessment could be performed using simulated reads.[75][76]
A note on assembly quality: The current consensus is that 1) assembly quality can vary depending on which metric is used, 2) assembly tools that scored well in one species do not necessarily perform well in the other species, and 3) combining different approaches might be the most reliable.[77][78][79]
Gene expression quantification
[edit]Expression is quantified to study cellular changes in response to external stimuli, differences between healthy and diseased states, and other research questions. Transcript levels are often used as a proxy for protein abundance, but these are often not equivalent due to post transcriptional events such as RNA interference and nonsense-mediated decay.[80]
Экспрессию определяют количественно путем подсчета количества прочтений, сопоставленных с каждым локусом на этапе сборки транскриптома . Экспрессию экзонов или генов можно оценить количественно с использованием контигов или аннотаций эталонных транскриптов. [10] Эти наблюдаемые количества считываний RNA-Seq были надежно подтверждены более старыми технологиями, включая экспрессионные микрочипы и qPCR . [57] [81] Инструменты, определяющие количество: HTSeq, [82] Количество функций, [83] Раунт, [84] максимальное количество, [85] ФИКСЕК, [86] и Каффвант. Эти инструменты определяют количество чтений на основе выровненных данных RNA-Seq, но подсчеты без выравнивания также можно получить с помощью Sailfish. [87] и Каллисто. [88] Затем количество прочтений преобразуется в соответствующие показатели для проверки гипотез, регрессии и другого анализа. Параметры для этого преобразования:
- Глубина/охват секвенирования . Хотя глубина секвенирования заранее указывается при проведении нескольких экспериментов по секвенированию РНК, она все равно будет сильно различаться между экспериментами. [89] Таким образом, общее количество чтений, сгенерированных в одном эксперименте, обычно нормализуется путем преобразования количества во фрагменты, чтения или количества на миллион сопоставленных чтений (FPM, RPM или CPM). Разница между RPM и FPM исторически возникла в ходе эволюции от одноконцевого секвенирования фрагментов к парному секвенированию. При одноконцевом секвенировании на каждый фрагмент выполняется только одно чтение ( т. е . RPM = FPM). При секвенировании парных концов на каждый фрагмент приходится два чтения ( т. е . RPM = 2 x FPM). Глубину секвенирования иногда называют размером библиотеки — количеством промежуточных молекул кДНК в эксперименте.
- Длина гена: более длинные гены будут иметь больше фрагментов/чтений/счетов, чем более короткие гены, если экспрессия транскриптов одинакова. Это корректируется путем деления FPM на длину признака (который может быть геном, транскриптом или экзоном), в результате чего получается количество метрических фрагментов на тысячу оснований признака на миллион картированных чтений (FPKM). [90] При просмотре групп функций в выборках FPKM преобразуется в количество транскриптов на миллион (TPM) путем деления каждой FPKM на сумму FPKM в выборке. [91] [92] [93]
- Общий выход РНК образца: поскольку из каждого образца извлекается одинаковое количество РНК, образцы с большим количеством общей РНК будут содержать меньше РНК на ген. Эти гены, по-видимому, имеют пониженную экспрессию, что приводит к ложноположительным результатам последующих анализов. [89] Стратегии нормализации, включая квантиль, DESeq2, TMM и медианное соотношение, пытаются учесть эту разницу путем сравнения набора недифференцированно экспрессируемых генов между образцами и соответствующего масштабирования. [94]
- Дисперсия экспрессии каждого гена: моделируется для учета ошибки выборки (важно для генов с низким количеством считываний), увеличения мощности и уменьшения ложноположительных результатов. Дисперсию можно оценить как нормальное , Пуассоновое или отрицательное биномиальное распределение. [95] [96] [97] и часто разлагается на технические и биологические вариации.
Всплески для абсолютного количественного определения и обнаружения общегеномных эффектов
[ редактировать ]Вставки РНК представляют собой образцы РНК в известных концентрациях, которые можно использовать в качестве золотых стандартов при разработке экспериментов и во время последующих анализов для абсолютного количественного определения и обнаружения общегеномных эффектов.
- Абсолютная количественная оценка: Абсолютная количественная оценка экспрессии генов невозможна в большинстве экспериментов RNA-Seq, которые количественно определяют экспрессию относительно всех транскриптов. Это возможно путем выполнения RNA-Seq с добавлением образцов РНК в известных концентрациях. После секвенирования количество считываний вставных последовательностей используется для определения взаимосвязи между количеством считываний каждого гена и абсолютным количеством биологических фрагментов. [13] [98] В одном примере этот метод использовали на эмбрионах Xenopus тропического для определения кинетики транскрипции. [99]
- Обнаружение общегеномных эффектов: изменения в глобальных регуляторах, включая ремоделеры хроматина , факторы транскрипции (например, MYC ), ацетилтрансферазные комплексы и расположение нуклеосом, не соответствуют предположениям о нормализации, и контроль всплеска может предложить точную интерпретацию. [100] [101]
Дифференциальное выражение
[ редактировать ]Самое простое, но зачастую наиболее эффективное применение RNA-Seq — это обнаружение различий в экспрессии генов между двумя или более состояниями ( например , леченными и нелеченными); этот процесс называется дифференциальным выражением. Выходные данные часто называют дифференциально экспрессируемыми генами (DEG), и эти гены могут иметь повышенную или пониженную регуляцию ( т. е . повышенную или пониженную регуляцию в интересующем состоянии). Существует множество инструментов, выполняющих дифференциальное выражение . Большинство из них запускаются в R , Python или командной строке Unix . Обычно используемые инструменты включают DESeq, [96] крайР, [97] и воом+лимма, [95] [102] все это доступно через R/ Bioconductor . [103] [104] Вот общие соображения при выполнении дифференциального выражения:
- Входные данные: входные данные для дифференциальной экспрессии включают (1) матрицу экспрессии RNA-Seq (M генов x N образцов) и (2) матрицу дизайна, содержащую экспериментальные условия для N образцов. Простейшая матрица проектирования содержит один столбец, соответствующий меткам проверяемого условия. Другие ковариаты (также называемые факторами, признаками, метками или параметрами) могут включать пакетные эффекты , известные артефакты и любые метаданные, которые могут искажать или опосредовать экспрессию генов. Помимо известных ковариат, неизвестные ковариаты также могут быть оценены с помощью подходов неконтролируемого машинного обучения , включая главный компонент , суррогатную переменную, [105] и ПИР [58] анализы. Анализ скрытых переменных часто используется для данных секвенирования РНК тканей человека, которые обычно содержат дополнительные артефакты, не отраженные в метаданных ( например , время ишемии, источники из нескольких учреждений, основные клинические характеристики, сбор данных за многие годы с участием большого количества сотрудников).
- Методы: Большинство инструментов используют регрессию или непараметрическую статистику для идентификации дифференциально экспрессируемых генов и основаны либо на количестве чтений, сопоставленных с эталонным геномом (DESeq2, limma, EdgeR), либо на основе количества чтений, полученных в результате количественного анализа без выравнивания (sleuth, [106] Манжета, [107] бальное платье [108] ). [109] После регрессии большинство инструментов используют корректировки p-значения либо коэффициента семейных ошибок (FWER) , либо коэффициента ложных открытий (FDR) для учета нескольких гипотез (в исследованиях на людях ~ 20 000 генов, кодирующих белок, или ~ 50 000 биотипов).
- Выходные данные: Типичные выходные данные состоят из строк, соответствующих количеству генов, и как минимум трех столбцов, логарифмического изменения каждого гена ( логарифмическое преобразование соотношения экспрессии между условиями, меры размера эффекта ), p-значения и p. -значение скорректировано для множественных сравнений . Гены считаются биологически значимыми, если они проходят пороговые значения по величине эффекта (логарифмическое изменение) и статистической значимости . В идеале эти пороговые значения должны быть указаны заранее , но характер экспериментов по секвенированию РНК часто носит исследовательский характер, поэтому трудно заранее предсказать размеры эффекта и соответствующие пороговые значения.
- Подводные камни: Смысл существования этих сложных методов состоит в том, чтобы избежать множества ловушек, которые могут привести к статистическим ошибкам и вводящим в заблуждение интерпретациям. Ловушки включают повышенный уровень ложноположительных результатов (из-за множественных сравнений), артефакты при подготовке образцов, гетерогенность образцов (например, смешанный генетический фон), сильно коррелированные образцы, неучтенные многоуровневые экспериментальные планы и плохой экспериментальный дизайн . Одной из примечательных ошибок является просмотр результатов в Microsoft Excel без использования функции импорта, чтобы гарантировать, что имена генов останутся текстовыми. [110] Несмотря на удобство, Excel автоматически преобразует некоторые имена генов ( SEPT1 , DEC1 , MARCH2 ) в даты или числа с плавающей запятой.
- Выбор инструментов и сравнительное тестирование: существует множество попыток сравнить результаты этих инструментов, причем DESeq2 имеет тенденцию умеренно превосходить другие методы. [111] [112] [113] [114] [19] [109] [115] [116] Как и другие методы, бенчмаркинг заключается в сравнении результатов инструмента друг с другом и с известными золотыми стандартами .
Последующий анализ списка дифференциально экспрессируемых генов бывает двух видов: проверка наблюдений и создание биологических выводов. Из-за ошибок дифференциальной экспрессии и секвенирования РНК важные наблюдения повторяются с помощью (1) ортогонального метода в тех же образцах (например, ПЦР в реальном времени ) или (2) другого, иногда заранее зарегистрированного , эксперимента в новой когорте. . Последнее помогает обеспечить возможность обобщения и обычно может сопровождаться метаанализом всех объединенных когорт. Наиболее распространенным методом получения биологического понимания результатов более высокого уровня является анализ обогащения набора генов , хотя иногда используются подходы с использованием генов-кандидатов. Обогащение набора генов определяет, является ли перекрытие между двумя наборами генов статистически значимым, в данном случае перекрытие между дифференциально экспрессируемыми генами и наборами генов из известных путей/баз данных ( например , Онтология генов , KEGG , Онтология фенотипа человека ) или из дополнительных анализов в одни и те же данные (например, сети совместного выражения). Общие инструменты для обогащения набора генов включают веб-интерфейсы ( например , ENRICHR, g:profiler, WEBGESTALT) [117] и пакеты программного обеспечения. При оценке результатов обогащения одна из эвристик заключается в том, чтобы сначала искать обогащение известной биологии в качестве проверки работоспособности, а затем расширять область поиска для поиска новой биологии.
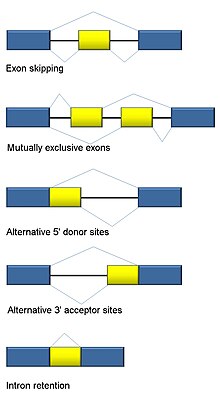
Альтернативный сплайсинг
[ редактировать ]Сплайсинг РНК является неотъемлемой частью эукариот и вносит значительный вклад в регуляцию и разнообразие белков, встречаясь более чем в 90% генов человека. [118] Существует несколько альтернативных режимов сплайсинга : пропуск экзонов (наиболее распространенный способ сплайсинга у людей и высших эукариот), взаимоисключающие экзоны, альтернативные донорные или акцепторные сайты, удержание интрона (наиболее распространенный способ сплайсинга у растений, грибов и простейших), альтернативное начало транскрипции. сайт (промотор) и альтернативное полиаденилирование. [118] Одна из целей RNA-Seq — идентифицировать альтернативные события сплайсинга и проверить, различаются ли они в разных условиях. Секвенирование длинного считывания фиксирует полный транскрипт и, таким образом, сводит к минимуму многие проблемы при оценке численности изоформ, такие как неоднозначное картирование чтения. Для короткого считывания RNA-Seq существует несколько методов обнаружения альтернативного сплайсинга, которые можно разделить на три основные группы: [119] [91] [120]
- На основе подсчета (также на основе событий, дифференциального сплайсинга): оценка удержания экзонов. Примеры: DEXSeq, [121] МАТС, [122] и SeqGSEA. [123]
- На основе изоформ (также модули многократного чтения, дифференциальное выражение изоформ) : сначала оценивают численность изоформ, а затем относительную численность между условиями. Примеры: Запонки 2. [124] и ДиффСплайс. [125]
- На основе вырезания интрона: расчет альтернативного сплайсинга с использованием разделенных чтений. Примеры: MAJIQ [126] и Листорез. [120]
Инструменты дифференциальной экспрессии генов также можно использовать для дифференциальной экспрессии изоформ, если изоформы заранее определены количественно с помощью других инструментов, таких как RSEM. [127]
Сети коэкспрессии
[ редактировать ]Сети коэкспрессии — это полученные на основе данных представления генов, которые ведут себя одинаково в разных тканях и экспериментальных условиях. [128] Их основная цель заключается в создании гипотез и подходах на основе ассоциации с виной для вывода о функциях ранее неизвестных генов. [128] Данные RNA-Seq использовались для вывода о генах, участвующих в определенных путях, на основе корреляции Пирсона , как у растений, так и у растений. [129] и млекопитающие. [130] Основным преимуществом данных RNA-Seq в этом виде анализа перед платформами микрочипов является способность охватить весь транскриптом, что дает возможность раскрыть более полные представления о регуляторных сетях генов. Дифференциальную регуляцию сплайсинговых изоформ одного и того же гена можно обнаружить и использовать для прогнозирования их биологических функций. [131] [132] Анализ сети взвешенной совместной экспрессии генов успешно использовался для идентификации модулей совместной экспрессии и внутримодульных генов-концентраторов на основе данных секвенирования РНК. Модули совместной экспрессии могут соответствовать типам клеток или путям. Внутримодульные концентраторы с высокой степенью связи можно интерпретировать как представителей соответствующего модуля. Собственный ген — это взвешенная сумма экспрессии всех генов в модуле. Собственные гены являются полезными биомаркерами (признаками) для диагностики и прогноза. [133] Были предложены подходы к стабилизирующему дисперсию преобразованию для оценки коэффициентов корреляции на основе данных секвенирования РНК. [129]
Открытие вариантов
[ редактировать ]RNA-Seq фиксирует вариации ДНК, включая однонуклеотидные варианты , небольшие вставки/делеции . и структурные вариации . Вызов вариантов в RNA-Seq аналогичен вызову вариантов ДНК и часто использует те же инструменты (включая SAMtools mpileup [134] и GATK HaplotypeCaller [135] ) с поправками для учета сращивания. Одним из уникальных аспектов вариантов РНК является аллель-специфическая экспрессия (ASE) : варианты только одного гаплотипа могут экспрессироваться преимущественно из-за регуляторных эффектов, включая импринтинг и экспрессию локусов количественных признаков , а также некодирующих редких вариантов . [136] [137] Ограничения идентификации вариантов РНК включают то, что она отражает только экспрессированные области (у людей <5% генома) и может быть подвержена систематическим ошибкам, вызванным обработкой данных (например, сборки транскриптома de novo недооценивают гетерозиготность [138] ), и имеет более низкое качество по сравнению с прямым секвенированием ДНК.
Редактирование РНК (посттранскрипционные изменения)
[ редактировать ]Наличие совпадающих геномных и транскриптомных последовательностей человека может помочь обнаружить посттранскрипционные изменения ( редактирование РНК ). [3] Событие посттранскрипционной модификации идентифицируется, если транскрипт гена имеет аллель/вариант, не наблюдаемый в геномных данных.
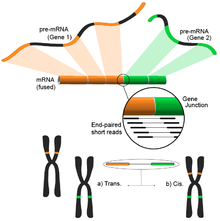
Обнаружение слитых генов
[ редактировать ]Вызванные различными структурными модификациями генома, слитые гены привлекли внимание из-за их связи с раком. [139] Способность RNA-Seq беспристрастно анализировать весь транскриптом образца делает его привлекательным инструментом для выявления подобных распространенных явлений при раке. [4]
Идея вытекает из процесса сопоставления коротких транскриптомных чтений с эталонным геномом. Большинство коротких ридов будут находиться в пределах одного полного экзона, а меньший, но все же большой набор, как ожидается, будет картироваться на известных соединениях экзон-экзон. Оставшиеся некартированные короткие чтения затем будут дополнительно проанализированы, чтобы определить, соответствуют ли они соединению экзон-экзон, где экзоны происходят из разных генов. Это было бы свидетельством возможного события слияния, однако из-за продолжительности считываний это может оказаться очень шумным. Альтернативный подход заключается в использовании парных чтений, когда потенциально большое количество парных ридов будет сопоставлять каждый конец с различным экзоном, обеспечивая лучшее освещение этих событий (см. рисунок). Тем не менее, конечный результат состоит из множества потенциально новых комбинаций генов, обеспечивающих идеальную отправную точку для дальнейшей проверки.
Копирование изменения номера
[ редактировать ]Анализ изменения числа копий (CNA) обычно используется в исследованиях рака. Приобретение и потеря генов имеют значение для сигнального пути и являются ключевым биомаркером молекулярной дисфункции в онкологии. Вызвать информацию CNA из данных RNA-Seq непросто из-за различий в экспрессии генов, которые приводят к разной величине различий в глубине считывания между генами. Из-за этих трудностей большая часть этих анализов обычно проводится с использованием полногеномного секвенирования/секвенирования всего экзома (WGS/WES). Но передовые инструменты биоинформатики могут вызывать CNA из RNA-Seq. [140]
Другие новые методы анализа и применения
[ редактировать ]Области применения RNA-Seq растут с каждым днем. Другое новое применение RNA-Seq включает обнаружение микробных примесей, [141] определение численности типов клеток (деконволюция типов клеток), [8] измерение экспрессии TE и предсказание неоантигена и т. д. [8]
История
[ редактировать ]
RNA-Seq был впервые разработан в середине 2000-х годов с появлением технологии секвенирования нового поколения. [144] Первые рукописи, в которых использовался RNA-Seq даже без использования этого термина, включают в себя исследования рака простаты. клеточных линий [145] (от 2006 г.), Medicago truncatula [146] (2006), кукуруза [147] (2007) и Arabidopsis thaliana [148] (2007), а сам термин «RNA-Seq» впервые был упомянут в 2008 году. [13] [149] Количество рукописей, упоминающих RNA-Seq в названии или аннотации (рисунок, синяя линия), постоянно увеличивается: в 2018 году было опубликовано 6754 рукописи. Пересечение RNA-Seq и медицины (рисунок, золотая линия) происходит с такой же скоростью. [150]
Приложения в медицине
[ редактировать ]RNA-Seq имеет потенциал для выявления новой биологии заболеваний, составления профиля биомаркеров для клинических показаний, определения путей воздействия лекарств и постановки генетических диагнозов. [151] [152] Эти результаты могут быть дополнительно персонализированы для подгрупп или даже отдельных пациентов, потенциально подчеркивая более эффективную профилактику, диагностику и терапию. Осуществимость этого подхода частично продиктована затратами денег и времени; связанным с этим ограничением является необходимость в команде специалистов (биоинформатиков, врачей/клиницистов, фундаментальных исследователей, технических специалистов) для полной интерпретации огромного количества данных, полученных в результате этого анализа. [153]
Масштабные усилия по секвенированию
[ редактировать ]Большое внимание было уделено данным RNA-Seq после того, как проекты «Энциклопедия элементов ДНК» (ENCODE) и «Атлас генома рака» (TCGA) использовали этот подход для характеристики десятков клеточных линий. [154] и тысячи образцов первичных опухолей, [155] соответственно. ENCODE нацелена на идентификацию полногеномных регуляторных областей в различных группах клеточных линий, и транскриптомные данные имеют первостепенное значение для понимания последующего эффекта этих эпигенетических и генетических регуляторных слоев. Вместо этого TCGA стремилась собрать и проанализировать тысячи образцов пациентов из 30 различных типов опухолей, чтобы понять основные механизмы злокачественной трансформации и прогрессирования. В этом контексте данные RNA-Seq дают уникальную картину транскриптомного статуса заболевания и рассматривают беспристрастную популяцию транскриптов, что позволяет идентифицировать новые транскрипты, слитые транскрипты и некодирующие РНК, которые можно было бы не обнаружить с помощью различных технологий.
См. также
[ редактировать ]Ссылки
[ редактировать ]![]() Эта статья была отправлена в WikiJournal of Science на внешнюю академическую рецензию в 2019 году ( отчеты рецензентов ). Обновленный контент был реинтегрирован на страницу Википедии по лицензии CC-BY-SA-3.0 ( 2021 г. ). Проверенная версия записи: Феликс Рихтер и др. (17 мая 2021 г.). «Общее введение в RNA-Seq» (PDF) . Викижурнал науки . 4 (2): 4. дои : 10.15347/WJS/2021.004 . ISSN 2470-6345 . Викиданные Q100146647 .
Эта статья была отправлена в WikiJournal of Science на внешнюю академическую рецензию в 2019 году ( отчеты рецензентов ). Обновленный контент был реинтегрирован на страницу Википедии по лицензии CC-BY-SA-3.0 ( 2021 г. ). Проверенная версия записи: Феликс Рихтер и др. (17 мая 2021 г.). «Общее введение в RNA-Seq» (PDF) . Викижурнал науки . 4 (2): 4. дои : 10.15347/WJS/2021.004 . ISSN 2470-6345 . Викиданные Q100146647 .
- ^ Лоу Р., Ширли Н., Бликли М., Долан С., Шафи Т. (май 2017 г.). «Технологии транскриптомики» . PLOS Вычислительная биология . 13 (5): e1005457. Бибкод : 2017PLSCB..13E5457L . дои : 10.1371/journal.pcbi.1005457 . ПМЦ 5436640 . ПМИД 28545146 .
- ^ Чу Ю, Кори Д.Р. (август 2012 г.). «Секвенирование РНК: выбор платформы, дизайн эксперимента и интерпретация данных» . Нуклеиновая кислотная терапия . 22 (4): 271–4. дои : 10.1089/нат.2012.0367 . ПМК 3426205 . ПМИД 22830413 .
- ^ Перейти обратно: а б с Ван З., Герштейн М., Снайдер М. (январь 2009 г.). «RNA-Seq: революционный инструмент для транскриптомики» . Обзоры природы. Генетика . 10 (1): 57–63. дои : 10.1038/nrg2484 . ПМЦ 2949280 . ПМИД 19015660 .
- ^ Перейти обратно: а б Махер К.А., Кумар-Синха С., Цао Х, Кальяна-Сундарам С., Хан Б., Цзин Х и др. (март 2009 г.). «Секвенирование транскриптома для обнаружения слияний генов при раке» . Природа . 458 (7234): 97–101. Бибкод : 2009Natur.458...97M . дои : 10.1038/nature07638 . ПМК 2725402 . ПМИД 19136943 .
- ^ Инголия НТ, Брар Г.А., Рускин С., Макгичи А.М., Вайсман Дж.С. (июль 2012 г.). «Стратегия профилирования рибосом для мониторинга трансляции in vivo путем глубокого секвенирования фрагментов мРНК, защищенных рибосомами» . Протоколы природы . 7 (8): 1534–50. дои : 10.1038/nprot.2012.086 . ПМК 3535016 . ПМИД 22836135 .
- ^ Альперн Д., Гардо В., Рассел Дж., Манжеат Б., Мейрелеш-Фильо А.С., Брейсс Р. и др. (апрель 2019 г.). «BRB-seq: сверхдоступная высокопроизводительная транскриптомика, обеспечиваемая массовым штрих-кодированием и секвенированием РНК» . Геномная биология . 20 (1): 71. дои : 10.1186/s13059-019-1671-x . ПМК 6474054 . ПМИД 30999927 .
- ^ Ли Дж.Х., Догарти Э.Р., Шейман Дж., Калхор Р., Ян Дж.Л., Ферранте Т.С. и др. (март 2014 г.). «Высокомультиплексное секвенирование субклеточной РНК in situ» . Наука . 343 (6177): 1360–3. Бибкод : 2014Sci...343.1360L . дои : 10.1126/science.1250212 . ПМК 4140943 . ПМИД 24578530 .
- ^ Перейти обратно: а б с Тинд А.С., Монга И., Тхакур П.К., Кумари П., Диндория К., Крзак М. и др. (ноябрь 2021 г.). «Демистификация новых приложений массового секвенирования РНК: применение и полезность биоинформатической методологии». Брифинги по биоинформатике . 22 (6). дои : 10.1093/нагрудник/bbab259 . ПМИД 34329375 .
- ^ Кукурба К.Р., Монтгомери С.Б. (апрель 2015 г.). «Секвенирование и анализ РНК» . Протоколы Колд-Спринг-Харбора . 2015 (11): 951–69. дои : 10.1101/pdb.top084970 . ПМЦ 4863231 . ПМИД 25870306 .
- ^ Перейти обратно: а б с д и Гриффит М., Уокер-младший, Spies NC, Эйнскоу Б.Дж., Гриффит О.Л. (август 2015 г.). «Информатика для секвенирования РНК: веб-ресурс для анализа в облаке» . PLOS Вычислительная биология . 11 (8): e1004393. Бибкод : 2015PLSCB..11E4393G . дои : 10.1371/journal.pcbi.1004393 . ПМЦ 4527835 . ПМИД 26248053 .
- ^ «РНК-секлопедия» . rnaseq.uoregon.edu . Проверено 8 февраля 2017 г.
- ^ Морин Р., Бейнбридж М., Фейес А., Херст М., Кшивински М., Пью Т. и др. (июль 2008 г.). «Профилирование транскриптома HeLa S3 с использованием случайно выбранной кДНК и массово-параллельного секвенирования с коротким считыванием» . БиоТехники . 45 (1): 81–94. дои : 10.2144/000112900 . ПМИД 18611170 .
- ^ Перейти обратно: а б с д Мортазави А., Уильямс Б.А., МакКью К., Шеффер Л., Уолд Б. (июль 2008 г.). «Картирование и количественная оценка транскриптомов млекопитающих с помощью RNA-Seq». Природные методы . 5 (7): 621–8. дои : 10.1038/nmeth.1226 . ПМИД 18516045 . S2CID 205418589 .
- ^ Сунь Ц, Хао Ц, Прасант К.В. (февраль 2018 г.). «Ядерные длинные некодирующие РНК: ключевые регуляторы экспрессии генов» . Тенденции в генетике . 34 (2): 142–157. дои : 10.1016/j.tig.2017.11.005 . ПМК 6002860 . ПМИД 29249332 .
- ^ Сигургерссон Б., Эмануэльссон О., Лундеберг Дж. (2014). «Секвенирование деградированной РНК, адресованной путем подсчета 3'-тегов» . ПЛОС ОДИН . 9 (3): е91851. Бибкод : 2014PLoSO...991851S . дои : 10.1371/journal.pone.0091851 . ПМЦ 3954844 . ПМИД 24632678 .
- ^ Чен Е.А., Суайая Т., Херштейн Дж.С., Евграфов О.В., Спицына В.Н., Реболини Д.Ф. и др. (октябрь 2014 г.). «Влияние целостности РНК на уникально картированные чтения в RNA-Seq» . Исследовательские заметки BMC . 7 : 753. doi : 10.1186/1756-0500-7-753 . ПМЦ 4213542 . ПМИД 25339126 .
- ^ Молл П., Анте М., Зейтц А., Реда Т. (декабрь 2014 г.). «Секвенирование мРНК QuantSeq 3' для количественного определения РНК». Природные методы . 11 (12): i – iii. дои : 10.1038/nmeth.f.376 . ISSN 1548-7105 . S2CID 83424788 .
- ^ Ойкономопулос С., Байега А., Фахиминия С., Джамбазиан Х., Берубе П., Рагуссис Дж. (2020). «Методики профилирования транскриптов с использованием давно известных технологий» . Границы генетики . 11 : 606. дои : 10.3389/fgene.2020.00606 . ПМЦ 7358353 . ПМИД 32733532 .
- ^ Перейти обратно: а б Конеса А., Мадригал П., Таразона С., Гомес-Кабреро Д., Сервера А., Макферсон А. и др. (январь 2016 г.). «Обзор лучших практик анализа данных секвенирования РНК» . Геномная биология . 17 (1): 13. дои : 10.1186/s13059-016-0881-8 . ПМЦ 4728800 . ПМИД 26813401 .
- ^ Лю Д., Грабер Дж. Х. (февраль 2006 г.). «Количественное сравнение библиотек EST требует компенсации систематических ошибок в генерации кДНК» . БМК Биоинформатика . 7:77 . дои : 10.1186/1471-2105-7-77 . ПМЦ 1431573 . ПМИД 16503995 .
- ^ Перейти обратно: а б с Гаральде Д.Р., Снелл Э.А., Яхимович Д., Сипос Б., Ллойд Дж.Х., Брюс М. и др. (март 2018 г.). «Высокопараллельное прямое секвенирование РНК на массиве нанопор». Природные методы . 15 (3): 201–206. дои : 10.1038/nmeth.4577 . ПМИД 29334379 . S2CID 3589823 .
- ^ Лю Д., Грабер Дж. Х. (февраль 2006 г.). «Количественное сравнение библиотек EST требует компенсации систематических ошибок в генерации кДНК» . БМК Биоинформатика . 7:77 . дои : 10.1186/1471-2105-7-77 . ПМЦ 1431573 . ПМИД 16503995 .
- ^ Глисон Дж., Лейн Т.А., Харрисон П.Дж., Харти В., Кларк М.Б. (3 августа 2020 г.). «Прямое секвенирование РНК нанопорами обнаруживает дифференциальную экспрессию между популяциями клеток человека» . bioRxiv : 2020.08.02.232785. дои : 10.1101/2020.08.02.232785 . S2CID 220975367 .
- ^ Перейти обратно: а б " Шапиро Э., Бизунер Т., Линнарссон С. (сентябрь 2013 г.). «Технологии, основанные на секвенировании отдельных клеток, произведут революцию в науке о целом организме». Обзоры природы. Генетика . 14 (9): 618–30. дои : 10.1038/nrg3542 . ПМИД 23897237 . S2CID 500845 . "
- ^ Колодзейчик А.А., Ким Дж.К., Свенссон В., Мариони Дж.К., Тейхманн С.А. (май 2015 г.). «Технология и биология секвенирования одноклеточных РНК» . Молекулярная клетка . 58 (4): 610–20. doi : 10.1016/j.molcel.2015.04.005 . ПМИД 26000846 .
- ^ Монторо Д.Т., Хабер А.Л., Битон М., Винарский В., Лин Б., Биркет С.Е. и др. (август 2018 г.). «Пересмотренная иерархия эпителия дыхательных путей включает ионоциты, экспрессирующие CFTR» . Природа . 560 (7718): 319–324. Бибкод : 2018Natur.560..319M . дои : 10.1038/s41586-018-0393-7 . ПМК 6295155 . ПМИД 30069044 .
- ^ Пласшерт Л.В., Жилионис Р., Чу-Винг Р., Савова В., Кнер Дж., Рома Г. и др. (август 2018 г.). «Одноклеточный атлас эпителия дыхательных путей обнаруживает богатые CFTR легочные ионоциты» . Природа . 560 (7718): 377–381. Бибкод : 2018Natur.560..377P . дои : 10.1038/s41586-018-0394-6 . ПМК 6108322 . ПМИД 30069046 .
- ^ Валиграч Л., Андрович П., Кубиста М. (март 2018 г.). «Платформы для сбора и анализа отдельных клеток» . Международный журнал молекулярных наук . 19 (3): 807. doi : 10.3390/ijms19030807 . ПМЦ 5877668 . ПМИД 29534489 .
- ^ Кляйн А.М., Мазутис Л., Акартуна И., Таллапрагада Н., Верес А., Ли В. и др. (май 2015 г.). «Капельное штрих-кодирование транскриптомики отдельных клеток применительно к эмбриональным стволовым клеткам» . Клетка . 161 (5): 1187–1201. дои : 10.1016/j.cell.2015.04.044 . ПМЦ 4441768 . ПМИД 26000487 .
- ^ Макоско Э.З., Басу А., Сатия Р., Немеш Дж., Шекхар К., Голдман М. и др. (май 2015 г.). «Высокопараллельное профилирование полногеномной экспрессии отдельных клеток с использованием нанолитровых капель» . Клетка . 161 (5): 1202–1214. дои : 10.1016/j.cell.2015.05.002 . ПМЦ 4481139 . ПМИД 26000488 .
- ^ Ислам С., Зейзель А., Йост С., Ла Манно Г., Заяк П., Каспер М. и др. (февраль 2014 г.). «Количественный секвенирование одноклеточной РНК с уникальными молекулярными идентификаторами». Природные методы . 11 (2): 163–6. дои : 10.1038/nmeth.2772 . ПМИД 24363023 . S2CID 6765530 .
- ^ " Хебенстрейт Д. (ноябрь 2012 г.). «Методы, проблемы и возможности секвенирования одноклеточной РНК» . Биология . 1 (3): 658–67. дои : 10.3390/biology1030658 . ПМК 4009822 . ПМИД 24832513 . "
- ^ Эбервин Дж., Сул Дж.Й., Бартфай Т., Ким Дж. (январь 2014 г.). «Обещание секвенирования отдельных клеток». Природные методы . 11 (1): 25–7. дои : 10.1038/nmeth.2769 . ПМИД 24524134 . S2CID 11575439 .
- ^ Тан Ф., Барбачору С., Ван Ю., Нордман Э., Ли С., Сюй Н. и др. (май 2009 г.). «Анализ всего транскриптома мРНК-Seq одной клетки». Природные методы . 6 (5): 377–82. дои : 10.1038/NMETH.1315 . ПМИД 19349980 . S2CID 16570747 .
- ^ Ислам С., Кьеллквист У., Молинер А., Заяц П., Фан Дж.Б., Лённерберг П. и др. (июль 2011 г.). «Характеристика одноклеточного транскрипционного ландшафта с помощью высокомультиплексного секвенирования РНК» . Геномные исследования . 21 (7): 1160–7. дои : 10.1101/гр.110882.110 . ПМЦ 3129258 . ПМИД 21543516 .
- ^ Рамшельд Д., Луо С., Ван Ю.К., Ли Р., Дэн К., Фаридани О.Р. и др. (август 2012 г.). «Полноразмерная мРНК-Seq из одноклеточных уровней РНК и отдельных циркулирующих опухолевых клеток» . Природная биотехнология . 30 (8): 777–82. дои : 10.1038/nbt.2282 . ПМЦ 3467340 . ПМИД 22820318 .
- ^ Хашимшони Т., Вагнер Ф., Шер Н., Янаи И. (сентябрь 2012 г.). «CEL-Seq: Seq одноклеточной РНК путем мультиплексной линейной амплификации» . Отчеты по ячейкам . 2 (3): 666–73. дои : 10.1016/j.celrep.2012.08.003 . ПМИД 22939981 .
- ^ Сингх М., Аль-Эриани Г., Карсвелл С., Фергюсон Дж. М., Блэкберн Дж., Бартон К. и др. (2018). «Высокопроизводительное целевое секвенирование отдельных клеток с длинным считыванием раскрывает клональный и транскрипционный ландшафт лимфоцитов» . биоRxiv . 10 (1): 3120. дои : 10.1101/424945 . ПМЦ 6635368 . ПМИД 31311926 .
- ^ Сасагава Ю., Никайдо И., Хаяси Т., Данно Х., Уно К.Д., Имаи Т. и др. (апрель 2013 г.). «Quartz-Seq: высоковоспроизводимый и чувствительный метод секвенирования одноклеточной РНК, выявляющий негенетическую гетерогенность экспрессии генов» . Геномная биология . 14 (4): С31. дои : 10.1186/gb-2013-14-4-r31 . ПМК 4054835 . ПМИД 23594475 .
- ^ Коуно Т., Муди Дж., Квон А.Т., Сибаяма Ю., Като С., Хуан Ю. и др. (январь 2019 г.). «C1 CAGE обнаруживает сайты начала транскрипции и активность энхансера с разрешением одной клетки» . Природные коммуникации . 10 (1): 360. Бибкод : 2019NatCo..10..360K . дои : 10.1038/s41467-018-08126-5 . ПМК 6341120 . ПМИД 30664627 .
- ^ Даль Молин А, Ди Камилло Б (2019). «Как разработать эксперимент по секвенированию одноклеточной РНК: подводные камни, проблемы и перспективы». Брифинги по биоинформатике . 20 (4): 1384–1394. дои : 10.1093/нагрудник/bby007 . ПМИД 29394315 .
- ^ Петерсон В.М., Чжан К.Х., Кумар Н., Вонг Дж., Ли Л., Уилсон Д.С. и др. (октябрь 2017 г.). «Мультиплексная количественная оценка белков и транскриптов в отдельных клетках». Природная биотехнология . 35 (10): 936–939. дои : 10.1038/nbt.3973 . ПМИД 28854175 . S2CID 205285357 .
- ^ Стоккиус М., Хафемейстер С., Стефенсон В., Хоук-Лумис Б., Чаттопадхай П.К., Свердлов Х. и др. (сентябрь 2017 г.). «Одновременное измерение эпитопа и транскриптома в отдельных клетках» . Природные методы . 14 (9): 865–868. дои : 10.1038/nmeth.4380 . ПМК 5669064 . ПМИД 28759029 .
- ^ Радж Б., Вагнер Д.Э., Маккенна А., Панди С., Кляйн А.М., Шендюр Дж. и др. (июнь 2018 г.). «Одновременное одноклеточное профилирование линий и типов клеток в мозге позвоночных» . Природная биотехнология . 36 (5): 442–450. дои : 10.1038/nbt.4103 . ПМЦ 5938111 . ПМИД 29608178 .
- ^ Олмос Д., Аркенау Х.Т., Анг Дж.Е., Ледаки И., Аттард Г., Карден К.П. и др. (январь 2009 г.). «Циркулирующие опухолевые клетки (ЦОК) считаются промежуточными конечными точками при кастрационно-резистентном раке предстательной железы (КРРПЖ): опыт одного центра» . Анналы онкологии . 20 (1): 27–33. дои : 10.1093/annonc/mdn544 . ПМИД 18695026 .
- ^ Левитин Х.М., Юань Дж., Симс П.А. (апрель 2018 г.). «Одноклеточный транскриптомный анализ гетерогенности опухоли» . Тенденции рака . 4 (4): 264–268. дои : 10.1016/j.trecan.2018.02.003 . ПМЦ 5993208 . ПМИД 29606308 .
- ^ Джерби-Арнон Л., Шах П., Куоко М.С., Родман С., Су М.Дж., Мелмс Дж.К. и др. (ноябрь 2018 г.). «Программа по борьбе с раковыми клетками способствует исключению Т-клеток и устойчивости к блокаде контрольных точек» . Клетка . 175 (4): 984–997.e24. дои : 10.1016/j.cell.2018.09.006 . ПМК 6410377 . ПМИД 30388455 .
- ^ Стивенсон В., Донлин Л.Т., Батлер А., Розо С., Бракен Б., Рашидфаррохи А. и др. (февраль 2018 г.). «Секвенирование одноклеточной РНК синовиальной ткани ревматоидного артрита с использованием недорогих микрофлюидных инструментов» . Природные коммуникации . 9 (1): 791. Бибкод : 2018NatCo...9..791S . дои : 10.1038/s41467-017-02659-x . ПМЦ 5824814 . ПМИД 29476078 .
- ^ Авраам Р., Хасли Н., Браун Д., Пенаранда С., Джиджон Х.Б., Тромбетта Дж.Дж. и др. (сентябрь 2015 г.). «Межклеточная изменчивость патогена приводит к гетерогенности иммунных реакций хозяина» . Клетка . 162 (6): 1309–21. дои : 10.1016/j.cell.2015.08.027 . ПМЦ 4578813 . ПМИД 26343579 .
- ^ Цао Дж., Пакер Дж.С., Рамани В., Кусанович Д.А., Хуинь С., Даза Р. и др. (август 2017 г.). «Комплексное одноклеточное транскрипционное профилирование многоклеточного организма» . Наука . 357 (6352): 661–667. Бибкод : 2017Sci...357..661C . дои : 10.1126/science.aam8940 . ПМЦ 5894354 . ПМИД 28818938 .
- ^ Пласс М., Солана Дж., Вольф Ф.А., Аюб С., Мисиос А., Глазар П. и др. (май 2018 г.). «Атлас типов клеток и древо происхождения целого сложного животного по данным одноклеточной транскриптомики» . Наука . 360 (6391): eaaq1723. дои : 10.1126/science.aaq1723 . ПМИД 29674432 .
- ^ Финчер К.Т., Вурцель О., де Хоог Т., Краварик К.М., Реддиен П.В. (май 2018 г.). «Средиземноморская Шмидтея» . Наука . 360 (6391): eaaq1736. дои : 10.1126/science.aaq1736 . ПМК 6563842 . ПМИД 29674431 .
- ^ Вагнер Д.Е., Вайнреб С., Коллинз З.М., Бриггс Дж.А., Мегасон С.Г., Кляйн А.М. (июнь 2018 г.). «Одноклеточное картирование ландшафтов экспрессии генов и их происхождения у эмбрионов рыбок данио» . Наука . 360 (6392): 981–987. Бибкод : 2018Sci...360..981W . дои : 10.1126/science.aar4362 . ПМК 6083445 . ПМИД 29700229 .
- ^ Фаррелл Дж.А., Ван Й., Ризенфельд С.Дж., Шекхар К., Регев А., Шир А.Ф. (июнь 2018 г.). «Одноклеточная реконструкция траекторий развития во время эмбриогенеза рыбок данио» . Наука . 360 (6392): eaar3131. дои : 10.1126/science.aar3131 . ПМК 6247916 . ПМИД 29700225 .
- ^ Бриггс Дж.А., Вайнреб С., Вагнер Д.Е., Мегасон С., Пешкин Л., Киршнер М.В. и др. (июнь 2018 г.). «Динамика экспрессии генов в эмбриогенезе позвоночных при разрешении отдельных клеток» . Наука . 360 (6392): eaar5780. дои : 10.1126/science.aar5780 . ПМК 6038144 . ПМИД 29700227 .
- ^ Ю Дж. «Научный прорыв 2018 года: отслеживание развития клетка за клеткой» . Научный журнал . Американская ассоциация содействия развитию науки.
- ^ Перейти обратно: а б Ли Х, Ловчи М.Т., Квон Ю.С., Розенфельд М.Г., Фу XD, Йео Г.В. (декабрь 2008 г.). «Определение плотности меток, необходимой для анализа цифрового транскриптома: применение к андроген-чувствительной модели рака простаты» . Труды Национальной академии наук Соединенных Штатов Америки . 105 (51): 20179–84. Бибкод : 2008PNAS..10520179L . дои : 10.1073/pnas.0807121105 . ПМК 2603435 . ПМИД 19088194 .
- ^ Перейти обратно: а б Стегл О, Партс Л, Пийпари М, Винн Дж, Дурбин Р (февраль 2012 г.). «Использование вероятностной оценки остатков экспрессии (PEER) для повышения мощности и интерпретируемости анализа экспрессии генов» . Протоколы природы . 7 (3): 500–7. дои : 10.1038/nprot.2011.457 . ПМЦ 3398141 . ПМИД 22343431 .
- ^ Кингсфорд С., Патро Р. (июнь 2015 г.). «Основное сжатие коротких последовательностей чтения с использованием кодирования пути» . Биоинформатика . 31 (12): 1920–8. doi : 10.1093/биоинформатика/btv071 . ПМЦ 4481695 . ПМИД 25649622 .
- ^ Перейти обратно: а б Грабхерр М.Г., Хаас Б.Дж., Яссур М., Левин Дж.З., Томпсон Д.А., Амит И. и др. (май 2011 г.). «Сборка полноразмерного транскриптома на основе данных RNA-Seq без эталонного генома» . Природная биотехнология . 29 (7): 644–52. дои : 10.1038/nbt.1883 . ПМЦ 3571712 . ПМИД 21572440 .
- ^ «Сборка De Novo с использованием чтения Illumina» (PDF) . Проверено 22 октября 2016 г. .
- ^ Оазисы: ассемблер транскриптома для очень коротких чтений.
- ^ Зербино Д.Р., Бирни Э. (май 2008 г.). «Бархат: алгоритмы сборки короткого чтения de novo с использованием графов де Брёйна» . Геномные исследования . 18 (5): 821–9. дои : 10.1101/гр.074492.107 . ПМК 2336801 . ПМИД 18349386 .
- ^ Чанг З., Ли Г., Лю Дж., Чжан Ю., Эшби С., Лю Д. и др. (февраль 2015 г.). «Bridger: новая основа для сборки транскриптома de novo с использованием данных RNA-seq» . Геномная биология . 16 (1): 30. дои : 10.1186/s13059-015-0596-2 . ПМЦ 4342890 . ПМИД 25723335 .
- ^ Бушманова Е, Антипов Д, Лапидус А, Пржибельский А.Д. (сентябрь 2019 г.). «rnaSPAdes: ассемблер транскриптома de novo и его применение к данным RNA-Seq» . ГигаСайенс . 8 (9). doi : 10.1093/gigascience/giz100 . ПМК 6736328 . ПМИД 31494669 .
- ^ Перейти обратно: а б Ли Б., Филлмор Н., Бай Ю., Коллинз М., Томсон Дж.А., Стюарт Р. и др. (декабрь 2014 г.). «Оценка сборок транскриптома de novo на основе данных RNA-Seq» . Геномная биология . 15 (12): 553. дои : 10.1186/s13059-014-0553-5 . ПМК 4298084 . ПМИД 25608678 .
- ^ Перейти обратно: а б Добин А., Дэвис К.А., Шлезингер Ф., Дренков Дж., Залески С., Джа С. и др. (январь 2013 г.). «STAR: сверхбыстрый универсальный выравниватель RNA-seq» . Биоинформатика . 29 (1): 15–21. doi : 10.1093/биоинформатика/bts635 . ПМК 3530905 . ПМИД 23104886 .
- ^ Лэнгмид Б. , Трапнелл С., Поп М., Зальцберг С.Л. (2009). «Сверхбыстрое и эффективное для памяти выравнивание коротких последовательностей ДНК с геномом человека» . Геномная биология . 10 (3): 25 рандов. дои : 10.1186/gb-2009-10-3-r25 . ПМК 2690996 . ПМИД 19261174 .
- ^ Трапнелл С., Пахтер Л. , Зальцберг С.Л. (май 2009 г.). «TopHat: обнаружение соединений сплайсинга с помощью RNA-Seq» . Биоинформатика . 25 (9): 1105–11. doi : 10.1093/биоинформатика/btp120 . ПМЦ 2672628 . ПМИД 19289445 .
- ^ Перейти обратно: а б Трапнелл С., Робертс А., Гофф Л., Пертеа Г., Ким Д., Келли Д.Р. и др. (март 2012 г.). «Дифференциальный анализ экспрессии генов и транскриптов в экспериментах по секвенированию РНК с помощью TopHat и Cufflinks» . Протоколы природы . 7 (3): 562–78. дои : 10.1038/нпрот.2012.016 . ПМЦ 3334321 . ПМИД 22383036 .
- ^ Ляо Ю., Смит Г.К., Ши В. (май 2013 г.). «Выравниватель Subread: быстрое, точное и масштабируемое сопоставление чтения путем посева и голосования» . Исследования нуклеиновых кислот . 41 (10): е108. дои : 10.1093/нар/gkt214 . ПМЦ 3664803 . ПМИД 23558742 .
- ^ Ким Д., Лэнгмид Б., Зальцберг С.Л. (апрель 2015 г.). «HISAT: выравниватель для быстрого сращивания с низкими требованиями к памяти» . Природные методы . 12 (4): 357–60. дои : 10.1038/nmeth.3317 . ПМЦ 4655817 . ПМИД 25751142 .
- ^ Ву Т.Д., Ватанабе С.К. (май 2005 г.). «GMAP: программа геномного картирования и выравнивания последовательностей мРНК и EST» . Биоинформатика . 21 (9): 1859–75. doi : 10.1093/биоинформатика/bti310 . ПМИД 15728110 .
- ^ Пертеа М., Пертеа ГМ, Антонеску КМ, Чанг ТК, Менделл Дж.Т., Зальцберг С.Л. (март 2015 г.). «StringTie позволяет улучшить реконструкцию транскриптома на основе считываний RNA-seq» . Природная биотехнология . 33 (3): 290–5. дои : 10.1038/nbt.3122 . ПМЦ 4643835 . ПМИД 25690850 .
- ^ Баруццо Дж., Хайер К.Е., Ким Э.Дж., Ди Камилло Б., Фитцджеральд Г.А., Грант Г.Р. (февраль 2017 г.). «Комплексный сравнительный анализ элайнеров RNA-seq на основе моделирования» . Природные методы . 14 (2): 135–139. дои : 10.1038/nmeth.4106 . ПМК 5792058 . ПМИД 27941783 .
- ^ Энгстрем П.Г., Стейгер Т., Сипос Б., Грант Г.Р., Калес А., Ретч Г. и др. (декабрь 2013 г.). «Систематическая оценка программ сплайсинга для данных секвенирования РНК» . Природные методы . 10 (12): 1185–91. дои : 10.1038/nmeth.2722 . ПМК 4018468 . ПМИД 24185836 .
- ^ Лу Б, Цзэн Цзи, Ши Т (февраль 2013 г.). «Сравнительное исследование стратегий сборки de novo и сборки на основе генома для реконструкции транскриптома на основе RNA-Seq» . Наука Китай Науки о жизни . 56 (2): 143–55. дои : 10.1007/s11427-013-4442-z . ПМИД 23393030 .
- ^ Брэднам К.Р., Фасс Дж.Н., Александров А., Баранай П., Бехнер М., Бироль И. и др. (июль 2013 г.). «Сборка 2: оценка методов сборки генома de novo у трех видов позвоночных» . ГигаСайенс . 2 (1): 10. arXiv : 1301.5406 . Бибкод : 2013arXiv1301.5406B . дои : 10.1186/2047-217X-2-10 . ПМЦ 3844414 . ПМИД 23870653 .
- ^ Хельцер М., Марц М. (май 2019 г.). «Сборка транскриптома de novo: всестороннее межвидовое сравнение ассемблеров короткого чтения RNA-Seq» . ГигаСайенс . 8 (5). doi : 10.1093/gigascience/giz039 . ПМК 6511074 . ПМИД 31077315 .
- ^ Гринбаум Д., Коланджело С., Уильямс К., Герштейн М. (2003). «Сравнение количества белков и уровней экспрессии мРНК в геномном масштабе» . Геномная биология . 4 (9): 117. doi : 10.1186/gb-2003-4-9-117 . ЧВК 193646 . ПМИД 12952525 .
- ^ Чжан З.Х., Джавери Д.Д., Маршалл В.М., Бауэр Д.С., Эдсон Дж., Нарайанан Р.К. и др. (август 2014 г.). «Сравнительное исследование методов дифференциального анализа экспрессии данных RNA-Seq» . ПЛОС ОДИН . 9 (8): е103207. Бибкод : 2014PLoSO...9j3207Z . дои : 10.1371/journal.pone.0103207 . ПМК 4132098 . ПМИД 25119138 .
- ^ Андерс С., Пил П.Т., Хубер В. (январь 2015 г.). «HTSeq — платформа Python для работы с данными высокопроизводительного секвенирования» . Биоинформатика . 31 (2): 166–9. doi : 10.1093/биоинформатика/btu638 . ПМК 4287950 . ПМИД 25260700 .
- ^ Ляо Ю., Смит Г.К., Ши В. (апрель 2014 г.). «featureCounts: эффективная программа общего назначения для присвоения считывания последовательностей геномным признакам». Биоинформатика . 30 (7): 923–30. arXiv : 1305.3347 . doi : 10.1093/биоинформатика/btt656 . ПМИД 24227677 .
- ^ Шмид М.В., Гроссниклаус У (февраль 2015 г.). «Rcount: простой и гибкий подсчет чтений RNA-Seq» . Биоинформатика . 31 (3): 436–7. doi : 10.1093/биоинформатика/btu680 . ПМИД 25322836 .
- ^ Финотелло Ф., Лавеццо Э., Бьянко Л., Барзон Л., Маццон П., Фонтана П. и др. (2014). «Уменьшение систематической ошибки в данных секвенирования РНК: новый подход к подсчету количества» . БМК Биоинформатика . 15 (Приложение 1): S7. дои : 10.1186/1471-2105-15-s1-s7 . ПМК 4016203 . ПМИД 24564404 .
- ^ Хасимото ТБ, Эдвардс, доктор медицины, Гиффорд Д.К. (март 2014 г.). «Универсальная коррекция счета для высокопроизводительного секвенирования» . PLOS Вычислительная биология . 10 (3): e1003494. Бибкод : 2014PLSCB..10E3494H . дои : 10.1371/journal.pcbi.1003494 . ПМЦ 3945112 . ПМИД 24603409 .
- ^ Патро Р., Маунт СМ, Кингсфорд К. (май 2014 г.). «Sailfish позволяет проводить количественный анализ изоформ без выравнивания на основе считываний РНК-seq с использованием облегченных алгоритмов» . Природная биотехнология . 32 (5): 462–4. arXiv : 1308.3700 . дои : 10.1038/nbt.2862 . ПМК 4077321 . ПМИД 24752080 .
- ^ Брэй Н.Л., Пиментел Х., Мелстед П., Пахтер Л. (май 2016 г.). «Почти оптимальная вероятностная количественная оценка секвенирования РНК» . Природная биотехнология . 34 (5): 525–7. дои : 10.1038/nbt.3519 . ПМИД 27043002 . S2CID 205282743 .
- ^ Перейти обратно: а б Робинсон, доктор медицинских наук, Ошлак А. (2010). «Метод нормализации масштабирования для анализа дифференциальной экспрессии данных секвенирования РНК» . Геномная биология . 11 (3): 25 р. дои : 10.1186/gb-2010-11-3-r25 . ПМЦ 2864565 . ПМИД 20196867 .
- ^ Трапнелл С., Уильямс Б.А., Пертеа Г., Мортазави А., Кван Г., ван Барен М.Дж. и др. (май 2010 г.). «Сборка транскриптов и количественная оценка с помощью RNA-Seq выявляют неаннотированные транскрипты и переключение изоформ во время дифференцировки клеток» . Природная биотехнология . 28 (5): 511–5. дои : 10.1038/nbt.1621 . ПМК 3146043 . ПМИД 20436464 .
- ^ Перейти обратно: а б Пахтер Л. (19 апреля 2011 г.). «Модели количественного определения транскриптов из RNA-Seq». arXiv : 1104.3889 [ q-bio.GN ].
- ^ «Что такое ФПКМ? Обзор единиц экспрессии RNA-Seq» . Фарраго . 8 мая 2014 года . Проверено 28 марта 2018 г.
- ^ Вагнер Г.П., Кин К., Линч В.Дж. (декабрь 2012 г.). «Измерение содержания мРНК с использованием данных секвенирования РНК: измерение RPKM непоследовательно в разных образцах». Теория в биологических науках . 131 (4): 281–5. дои : 10.1007/s12064-012-0162-3 . ПМИД 22872506 . S2CID 16752581 .
- ^ Эванс С., Хардин Дж., Стобель Д.М. (сентябрь 2018 г.). «Выбор методов нормализации межобразцовой РНК-Seq с точки зрения их предположений» . Брифинги по биоинформатике . 19 (5): 776–792. дои : 10.1093/нагрудник/bbx008 . ПМК 6171491 . ПМИД 28334202 .
- ^ Перейти обратно: а б Лоу К.В., Чен Ю, Ши В, Смит Г.К. (февраль 2014 г.). «voom: Прецизионные веса открывают инструменты анализа линейных моделей для подсчета считываний РНК-секвенирования» . Геномная биология . 15 (2): Р29. дои : 10.1186/gb-2014-15-2-r29 . ПМК 4053721 . ПМИД 24485249 .
- ^ Перейти обратно: а б Андерс С., Хубер В. (2010). «Дифференциальный экспрессионный анализ данных подсчета последовательностей» . Геномная биология . 11 (10): Р106. дои : 10.1186/gb-2010-11-10-r106 . ПМК 3218662 . ПМИД 20979621 .
- ^ Перейти обратно: а б Робинсон, доктор медицинских наук, Маккарти диджей, Смит Г.К. (январь 2010 г.). «edgeR: пакет Bioconductor для дифференциального анализа экспрессии цифровых данных об экспрессии генов» . Биоинформатика . 26 (1): 139–40. doi : 10.1093/биоинформатика/btp616 . ПМК 2796818 . ПМИД 19910308 .
- ^ Маргерат С., Шмидт А., Кодлин С., Чен В., Эберсольд Р., Бэлер Дж. (октябрь 2012 г.). «Количественный анализ транскриптомов и протеомов делящихся дрожжей в пролиферирующих и покоящихся клетках» . Клетка . 151 (3): 671–83. дои : 10.1016/j.cell.2012.09.019 . ПМЦ 3482660 . ПМИД 23101633 .
- ^ Оуэнс Н.Д., Блиц И.Л., Лейн М.А., Патрушев И., Овертон Дж.Д., Гилкрист М.Дж. и др. (январь 2016 г.). «Измерение абсолютного числа копий РНК с высоким временным разрешением показывает кинетику транскриптома в развитии» . Отчеты по ячейкам . 14 (3): 632–647. дои : 10.1016/j.celrep.2015.12.050 . ПМЦ 4731879 . ПМИД 26774488 .
- ^ Чен К., Ху Z, Ся Z, Чжао Д., Ли В., Тайлер Дж. К. (декабрь 2015 г.). «Упущенный факт: фундаментальная необходимость контроля всплесков практически для всех полногеномных анализов» . Молекулярная и клеточная биология . 36 (5): 662–7. дои : 10.1128/MCB.00970-14 . ПМК 4760223 . ПМИД 26711261 .
- ^ Ловен Дж., Орландо Д.А., Сигова А.А., Лин С.И., Рал П.Б., Бердж С.Б. и др. (октябрь 2012 г.). «Возвращаясь к глобальному анализу экспрессии генов» . Клетка . 151 (3): 476–82. дои : 10.1016/j.cell.2012.10.012 . ПМЦ 3505597 . ПМИД 23101621 .
- ^ Ричи М.Э., Фипсон Б., Ву Д., Ху Ю., Лоу К.В., Ши В. и др. (апрель 2015 г.). «Лимма обеспечивает дифференциальный анализ экспрессии для секвенирования РНК и исследований на микрочипах» . Исследования нуклеиновых кислот . 43 (7): е47. дои : 10.1093/нар/gkv007 . ПМК 4402510 . ПМИД 25605792 .
- ^ «Биокондуктор — программное обеспечение с открытым исходным кодом для биоинформатики» .
- ^ Хубер В., Кэри В.Дж., Джентльмен Р., Андерс С., Карлсон М., Карвалью Б.С. и др. (февраль 2015 г.). «Организация высокопроизводительного геномного анализа с помощью Bioconductor» . Природные методы . 12 (2): 115–21. дои : 10.1038/nmeth.3252 . ПМК 4509590 . ПМИД 25633503 .
- ^ Лик Дж.Т., Стори Дж.Д. (сентябрь 2007 г.). «Учет гетерогенности в исследованиях экспрессии генов с помощью анализа суррогатных переменных» . ПЛОС Генетика . 3 (9): 1724–35. дои : 10.1371/journal.pgen.0030161 . ЧВК 1994707 . ПМИД 17907809 .
- ^ Пиментел Х., Брэй Н.Л., Пуэнте С., Мелстед П., Пахтер Л. (июль 2017 г.). «Дифференциальный анализ РНК-секвенирования с учетом неопределенности количественного определения» . Природные методы . 14 (7): 687–690. дои : 10.1038/nmeth.4324 . ПМИД 28581496 . S2CID 15063247 .
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Дж.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq» . Природная биотехнология . 31 (1): 46–53. дои : 10.1038/nbt.2450 . ПМЦ 3869392 . ПМИД 23222703 .
- ^ Фрейзи А.С., Пертеа Дж., Джаффе А.Е., Лэнгмид Б., Зальцберг С.Л., Лик Дж.Т. (март 2015 г.). «Ballgown устраняет разрыв между сборкой транскриптома и анализом экспрессии» . Природная биотехнология . 33 (3): 243–6. дои : 10.1038/nbt.3172 . ПМЦ 4792117 . ПМИД 25748911 .
- ^ Перейти обратно: а б Сахрейян С.М., Мохиюддин М., Себра Р., Тилгнер Х., Афшар П.Т., Ау К.Ф. и др. (июль 2017 г.). «Получение всестороннего биологического понимания транскриптома путем проведения анализа секвенирования РНК широкого спектра» . Природные коммуникации . 8 (1): 59. Бибкод : 2017NatCo...8...59S . дои : 10.1038/s41467-017-00050-4 . ПМЦ 5498581 . ПМИД 28680106 .
- ^ Циманн М., Эрен Ю., Эль-Оста А. (август 2016 г.). «Ошибки в названиях генов широко распространены в научной литературе» . Геномная биология . 17 (1): 177. дои : 10.1186/s13059-016-1044-7 . ПМЦ 4994289 . ПМИД 27552985 .
- ^ Сонесон С., Делоренци М. (март 2013 г.). «Сравнение методов дифференциального анализа экспрессии данных секвенирования РНК» . БМК Биоинформатика . 14:91 . дои : 10.1186/1471-2105-14-91 . ПМК 3608160 . ПМИД 23497356 .
- ^ Фонсека Н.А., Мариони Дж., Бразма А. (30 сентября 2014 г.). «Профилирование генов RNA-Seq - систематическое эмпирическое сравнение» . ПЛОС ОДИН . 9 (9): e107026. Бибкод : 2014PLoSO...9j7026F . дои : 10.1371/journal.pone.0107026 . ПМЦ 4182317 . ПМИД 25268973 .
- ^ Сейеднасролла Ф., Лайхо А., Эло Л.Л. (январь 2015 г.). «Сравнение пакетов программного обеспечения для обнаружения дифференциальной экспрессии в исследованиях РНК-секвенирования» . Брифинги по биоинформатике . 16 (1): 59–70. дои : 10.1093/нагрудник/bbt086 . ПМЦ 4293378 . ПМИД 24300110 .
- ^ Рапапорт Ф., Ханин Р., Лян Ю., Пирун М., Крек А., Зумбо П. и др. (2013). «Комплексная оценка методов анализа дифференциальной экспрессии генов для данных секвенирования РНК» . Геномная биология . 14 (9): 95 рандов. дои : 10.1186/gb-2013-14-9-r95 . ПМЦ 4054597 . ПМИД 24020486 .
- ^ Коста-Сильва Х., Домингес Д., Лопес FM (21 декабря 2017 г.). «Анализ дифференциальной экспрессии RNA-Seq: расширенный обзор и программный инструмент» . ПЛОС ОДИН . 12 (12): e0190152. Бибкод : 2017PLoSO..1290152C . дои : 10.1371/journal.pone.0190152 . ПМЦ 5739479 . ПМИД 29267363 .
- ^ Корчете Л.А., Рохас Э.А., Алонсо-Лопес Д., Де Лас Ривас Х., Гутьеррес Н.К., Бургильо Ф.Д. (12 ноября 2020 г.). «Систематическое сравнение и оценка процедур RNA-seq для количественного анализа экспрессии генов» . Научные отчеты . 12 (10): 19737. Бибкод : 2020NatSR..1019737C . дои : 10.1038/s41598-020-76881-x . ПМЦ 7665074 . ПМИД 33184454 .
- ^ Ляо И., Ван Дж., Джениг Э.Дж., Ши З., Чжан Б. (июль 2019 г.). «WebGestalt 2019: набор инструментов для анализа набора генов с обновленными пользовательскими интерфейсами и API» . Исследования нуклеиновых кислот . 47 (П1): И199–205. дои : 10.1093/nar/gkz401 . ПМК 6602449 . ПМИД 31114916 .
- ^ Перейти обратно: а б Керен Х., Лев-Маор Г., Аст Г. (май 2010 г.). «Альтернативный сплайсинг и эволюция: диверсификация, определение экзонов и функции». Обзоры природы. Генетика . 11 (5): 345–55. дои : 10.1038/nrg2776 . ПМИД 20376054 . S2CID 5184582 .
- ^ Лю Р., Лорейн А.Э., Дикерсон Дж.А. (декабрь 2014 г.). «Сравнение вычислительных методов дифференциального обнаружения альтернативного сплайсинга с использованием RNA-seq в растительных системах» . БМК Биоинформатика . 15 (1): 364. дои : 10.1186/s12859-014-0364-4 . ПМК 4271460 . ПМИД 25511303 .
- ^ Перейти обратно: а б Ли Й.И., Ноулз Д.А., Хамфри Дж., Барбейра А.Н., Дикинсон С.П., Им Х.К. и др. (январь 2018 г.). «Количественная оценка сплайсинга РНК без аннотаций с использованием LeafCutter» . Природная генетика . 50 (1): 151–158. дои : 10.1038/s41588-017-0004-9 . ПМК 5742080 . ПМИД 29229983 .
- ^ Андерс С., Рейес А., Хубер В. (октябрь 2012 г.). «Обнаружение дифференциального использования экзонов на основе данных РНК-секвенирования» . Геномные исследования . 22 (10): 2008–17. дои : 10.1101/гр.133744.111 . ПМК 3460195 . ПМИД 22722343 .
- ^ Шен С., Парк Дж.В., Хуан Дж., Диттмар К.А., Лу ZX, Чжоу Q и др. (апрель 2012 г.). «MATS: байесовский подход для гибкого обнаружения дифференциального альтернативного сплайсинга на основе данных RNA-Seq» . Исследования нуклеиновых кислот . 40 (8): е61. дои : 10.1093/nar/gkr1291 . ПМЦ 3333886 . ПМИД 22266656 .
- ^ Ван X, MJ Кэрнс (июнь 2014 г.). «SeqGSEA: пакет Bioconductor для анализа обогащения набора генов данными RNA-Seq, объединяющий дифференциальную экспрессию и сплайсинг» . Биоинформатика . 30 (12): 1777–9. doi : 10.1093/биоинформатика/btu090 . ПМИД 24535097 .
- ^ Трапнелл С., Хендриксон Д.Г., Соважо М., Гофф Л., Ринн Дж.Л., Пахтер Л. (январь 2013 г.). «Дифференциальный анализ регуляции генов при разрешении транскриптов с помощью RNA-seq» . Природная биотехнология . 31 (1): 46–53. дои : 10.1038/nbt.2450 . ПМЦ 3869392 . ПМИД 23222703 .
- ^ Ху Ю, Хуан Ю, Ду Ю, Орельяна С.Ф., Сингх Д., Джонсон А.Р. и др. (январь 2013 г.). «DiffSplice: полногеномное обнаружение событий дифференциального сплайсинга с помощью RNA-seq» . Исследования нуклеиновых кислот . 41 (2): е39. дои : 10.1093/нар/gks1026 . ПМЦ 3553996 . ПМИД 23155066 .
- ^ Вакеро-Гарсия Дж., Баррера А., Газзара М.Р., Гонсалес-Валлинас Дж., Лаэнс Н.Ф., Хогенеш Дж.Б. и др. (февраль 2016 г.). «Новый взгляд на сложность и регуляцию транскриптома через призму локальных вариаций сплайсинга» . электронная жизнь . 5 : е11752. дои : 10.7554/eLife.11752 . ПМК 4801060 . ПМИД 26829591 .
- ^ Мерино Г.А., Конеса А., Фернандес Э.А. (март 2019 г.). «Сравнительный анализ рабочих процессов для обнаружения дифференциального сплайсинга и дифференциальной экспрессии на уровне изоформ в исследованиях секвенирования РНК человека». Брифинги по биоинформатике . 20 (2): 471–481. дои : 10.1093/нагрудник/bbx122 . hdl : 11336/41247 . ПМИД 29040385 . S2CID 22706028 .
- ^ Перейти обратно: а б Маркотт Э.М., Пеллегрини М., Томпсон М.Дж., Йейтс Т.О., Айзенберг Д. (ноябрь 1999 г.). «Комбинированный алгоритм полногеномного прогнозирования функции белка». Природа . 402 (6757): 83–6. Бибкод : 1999Natur.402...83M . дои : 10.1038/47048 . ПМИД 10573421 . S2CID 144447 .
- ^ Перейти обратно: а б Георгий Ф.М., Дель Фаббро С., Ликаузи Ф. (март 2013 г.). «Сравнительное исследование сетей коэкспрессии, полученных на основе РНК-секвенирования и микрочипов, у Arabidopsis thaliana» . Биоинформатика . 29 (6): 717–24. doi : 10.1093/биоинформатика/btt053 . HDL : 11390/990155 . ПМИД 23376351 .
- ^ Янку О.Д., Каване С., Боттомли Д., Сирлз Р., Хитземанн Р., МакВини С. (июнь 2012 г.). «Использование данных RNA-Seq для вывода о сети коэкспрессии de novo» . Биоинформатика . 28 (12): 1592–7. doi : 10.1093/биоинформатика/bts245 . ПМЦ 3493127 . ПМИД 22556371 .
- ^ Экси Р., Ли Х.Д., Менон Р., Вэнь Ю., Оменн Г.С., Крецлер М. и др. (ноябрь 2013 г.). «Систематическое дифференцирование функций альтернативно сплайсированных изоформ посредством интеграции данных секвенирования РНК» . PLOS Вычислительная биология . 9 (11): e1003314. Бибкод : 2013PLSCB...9E3314E . дои : 10.1371/journal.pcbi.1003314 . ПМЦ 3820534 . ПМИД 24244129 .
- ^ Ли Х.Д., Менон Р., Оменн Г.С., Гуань Ю (август 2014 г.). «Новая эра интеграции геномных данных для анализа функции изоформ сплайсинга» . Тенденции в генетике . 30 (8): 340–7. дои : 10.1016/j.tig.2014.05.005 . ПМЦ 4112133 . ПМИД 24951248 .
- ^ Форушани А., Аграхари Р., Докинг Р., Чанг Л., Дунс Г., Худоба М. и др. (март 2017 г.). «Крупномасштабный анализ генной сети показывает значение пути внеклеточного матрикса и гомеобоксных генов при остром миелолейкозе: введение в пакет Pigengene и его применение» . BMC Медицинская Геномика . 10 (1): 16. дои : 10.1186/s12920-017-0253-6 . ПМЦ 5353782 . ПМИД 28298217 .
- ^ Ли Х., Рукосакер Б., Вайсокер А., Феннелл Т., Руан Дж., Гомер Н. и др. (август 2009 г.). «Формат Sequence Alignment/Map и SAMtools» . Биоинформатика . 25 (16): 2078–9. doi : 10.1093/биоинформатика/btp352 . ПМК 2723002 . ПМИД 19505943 .
- ^ ДеПристо М.А., Бэнкс Э., Поплин Р., Гаримелла К.В., Магуайр Дж.Р., Хартл С. и др. (май 2011 г.). «Система обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК нового поколения» . Природная генетика . 43 (5): 491–8. дои : 10.1038/ng.806 . ПМК 3083463 . ПМИД 21478889 .
- ^ Battle A, Brown CD, Энгельхардт BE, Монтгомери SB (октябрь 2017 г.). «Генетическое влияние на экспрессию генов в тканях человека» . Природа . 550 (7675): 204–213. Бибкод : 2017Natur.550..204A . дои : 10.1038/nature24277 . hdl : 10230/34202 . ПМК 5776756 . ПМИД 29022597 .
- ^ Рихтер Ф., Хоффман Г.Э., Манхаймер К.Б., Патель Н., Шарп А.Дж., МакКин Д. и др. (октябрь 2019 г.). «ORE определяет крайние эффекты экспрессии, обогащенные редкими вариантами» . Биоинформатика . 35 (20): 3906–3912. doi : 10.1093/биоинформатика/btz202 . ПМК 6792115 . ПМИД 30903145 .
- ^ Фридман А.Х., Клэмп М., Сактон ТБ (январь 2021 г.). «Ошибка, шум и предвзятость в сборках транскриптома de novo». Ресурсы молекулярной экологии . 21 (1): 18–29. дои : 10.1111/1755-0998.13156 . ПМИД 32180366 . S2CID 212739959 .
- ^ Тейшейра М.Р. (декабрь 2006 г.). «Рецидивирующие онкогены слияния при карциномах». Критические обзоры онкогенеза . 12 (3–4): 257–71. дои : 10.1615/critrevoncog.v12.i3-4.40 . ПМИД 17425505 . S2CID 40770452 .
- ^ Тинд А.С., Монга И., Тхакур П.К., Кумари П., Диндория К., Крзак М. и др. (ноябрь 2021 г.). «Демистификация новых приложений массового секвенирования РНК: применение и полезность биоинформатической методологии». Брифинги по биоинформатике . 22 (6). дои : 10.1093/нагрудник/bbab259 . ПМИД 34329375 .
- ^ Санджованни М., Граната И., Тинд А.С., Гуаррачино М.Р. (апрель 2019 г.). «От мусора к сокровищу: обнаружение неожиданного загрязнения в некартированных данных NGS» . БМК Биоинформатика . 20 (Приложение 4): 168. doi : 10.1186/s12859-019-2684-x . ПМК 6472186 . ПМИД 30999839 .
- ^ «Поиск в PubMed: «RNA Seq» ИЛИ «RNA-Seq» ИЛИ «Секвенирование РНК» ИЛИ «RNASeq» » . ПабМед . Проверено 20 июня 2021 г.
- ^ «Поиск в PubMed: («RNA Seq» ИЛИ «RNA-Seq» ИЛИ «Секвенирование РНК» ИЛИ «RNASeq») И «Медицина» " . ПабМед . Проверено 20 июня 2021 г.
- ^ Вебер А.П. (ноябрь 2015 г.). «Открытие новой биологии посредством секвенирования РНК» . Физиология растений . 169 (3): 1524–31. дои : 10.1104/стр.15.01081 . ПМК 4634082 . ПМИД 26353759 .
- ^ Бейнбридж М.Н., Уоррен Р.Л., Херст М., Романуик Т., Зенг Т., Го А. и др. (сентябрь 2006 г.). «Анализ транскриптома линии клеток рака простаты LNCaP с использованием подхода секвенирования путем синтеза» . БМК Геномика . 7 : 246. дои : 10.1186/1471-2164-7-246 . ПМЦ 1592491 . ПМИД 17010196 .
- ^ Чунг Ф., Хаас Б.Дж., Голдберг С.М., Мэй Г.Д., Сяо Ю., Town CD (октябрь 2006 г.). «Секвенирование Medicago truncatula экспрессировало секвенированные теги с использованием технологии 454 Life Sciences» . БМК Геномика . 7 : 272. дои : 10.1186/1471-2164-7-272 . ПМК 1635983 . ПМИД 17062153 .
- ^ Эмрих С.Дж., Барбазук В.Б., Ли Л., Шнабле П.С. (январь 2007 г.). «Открытие и аннотация генов с использованием секвенирования транскриптома LCM-454» . Геномные исследования . 17 (1): 69–73. дои : 10.1101/гр.5145806 . ПМК 1716268 . ПМИД 17095711 .
- ^ Вебер А.П., Вебер К.Л., Карр К., Вилкерсон С., Ольрогге Дж.Б. (май 2007 г.). «Отбор проб транскриптома Arabidopsis с помощью массового параллельного пиросеквенирования» . Физиология растений . 144 (1): 32–42. дои : 10.1104/стр.107.096677 . ЧВК 1913805 . ПМИД 17351049 .
- ^ Нагалакшми У., Ван З., Ваерн К., Шу С., Раха Д., Герштейн М. и др. (июнь 2008 г.). «Транскрипционный ландшафт генома дрожжей, определенный с помощью секвенирования РНК» . Наука . 320 (5881): 1344–9. Бибкод : 2008Sci...320.1344N . дои : 10.1126/science.1158441 . ПМЦ 2951732 . ПМИД 18451266 .
- ^ Рихтер Ф (2021). «Широкое введение в RNA-Seq» . Викижурнал науки . 4 (1): 4. doi : 10.15347/WJS/2021.004 .
- ^ Каммингс Б.Б., Маршалл Дж.Л., Тукиайнен Т., Лек М., Донкерворт С., Фоли А.Р. и др. (19 апреля 2017 г.). «Улучшение генетической диагностики менделевской болезни с помощью секвенирования транскриптома». Наука трансляционной медицины . 9 (386). doi : 10.1126/scitranslmed.aal5209 . hdl : 10230/35912 .
- ^ Кремер Л.С., Бадер Д.М., Мертес С., Копайтич Р., Пихлер Г., Юсо А. и др. (12 июня 2017 г.). «Генетическая диагностика менделевских расстройств с помощью секвенирования РНК». Природные коммуникации . 8 (1). дои : 10.1038/ncomms15824 .
- ^ Сандберг Р. (январь 2014 г.). «Вход в эпоху одноклеточной транскриптомики в биологии и медицине» . Природные методы . 11 (1): 22–4. дои : 10.1038/nmeth.2764 . ПМИД 24524133 . S2CID 27632439 .
- ^ «КОДИРОВАТЬ матрицу данных» . Проверено 28 июля 2013 г.
- ^ «Атлас генома рака - Портал данных» . Проверено 28 июля 2013 г.
Дальнейшее чтение
[ редактировать ]- Тагучи Ю. (2019). «Сравнительный транскриптомический анализ». Энциклопедия биоинформатики и вычислительной биологии . стр. 814–818. дои : 10.1016/B978-0-12-809633-8.20163-5 . ISBN 978-0-12-811432-2 . S2CID 65302519 .
Внешние ссылки
[ редактировать ]- Креско Б., Фёлкер Р., Смолл С. (2001). Башем С., Кэтчен Дж. (ред.). «РНК-секлопедия» . Университет Орегона. : общее руководство по планированию и проведению эксперимента по секвенированию РНК.