Последовательность нуклеиновой кислоты
Эта статья нуждается в дополнительных цитатах для проверки . ( март 2014 г. ) |
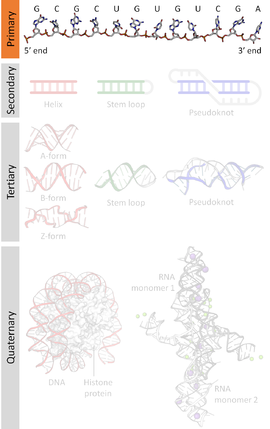
Последовательность нуклеиновой кислоты представляет собой последовательность оснований в нуклеотидах, образующих аллели внутри молекулы ДНК (с использованием GACT) или РНК (GACU). Эта последовательность обозначается серией из пяти разных букв, обозначающих порядок нуклеотидов. По соглашению последовательности обычно располагаются от 5'-конца к 3'-концу . Для ДНК с ее двойной спиралью существует два возможных направления обозначенной последовательности; из этих двух смысловая цепь используется . Поскольку нуклеиновые кислоты обычно представляют собой линейные (неразветвленные) полимеры , определение последовательности эквивалентно определению ковалентной структуры всей молекулы. По этой причине последовательность нуклеиновой кислоты также называют первичной структурой .
Последовательность представляет генетическую информацию . Биологическая дезоксирибонуклеиновая кислота представляет собой информацию , направляющую функции организма .
Нуклеиновые кислоты также имеют вторичную структуру и третичную структуру . Первичную структуру иногда ошибочно называют «первичной последовательностью». Однако параллельной концепции вторичной или третичной последовательности не существует.
Нуклеотиды [ править ]
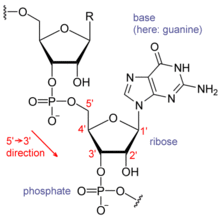

Нуклеиновые кислоты состоят из цепочки связанных единиц, называемых нуклеотидами. Каждый нуклеотид состоит из трех субъединиц: фосфатная группа и сахар ( рибоза в случае РНК , дезоксирибоза в ДНК ) составляют основу цепи нуклеиновой кислоты, а к сахару присоединено одно из множества нуклеиновых оснований . Нуклеиновые основания играют важную роль в спаривании оснований нитей для формирования вторичных и третичных структур более высокого уровня, таких как знаменитая двойная спираль .
Возможные буквы: A , C , G и T , обозначающие четыре нуклеотидных основания цепи ДНК – аденин , цитозин , гуанин , тимин – ковалентно связанные с фосфодиэфирным остовом. В типичном случае последовательности печатаются примыкающими друг к другу без промежутков, как в последовательности AAAGTCTGAC, читаемой слева направо в направлении от 5’ к 3’ . Что касается транскрипции , последовательность находится в кодирующей цепи, если она имеет тот же порядок, что и транскрибируемая РНК.
Одна последовательность может быть комплементарна другой последовательности, что означает, что они имеют основание в каждой позиции в комплементарном (т. е. от A до T, от C до G) и в обратном порядке. Например, последовательность, комплементарная TTAC, представляет собой GTAA. Если одна цепь двухцепочечной ДНК считается смысловой цепью, то другая цепь, называемая антисмысловой цепью, будет иметь последовательность, комплементарную смысловой цепи.
Обозначения [ править ]
Хотя A, T, C и G обозначают конкретный нуклеотид в определенной позиции, существуют также буквы, обозначающие неоднозначность, которые используются, когда в этой позиции может находиться более одного типа нуклеотидов. Правила Международного союза теоретической и прикладной химии ( IUPAC ) заключаются в следующем: [1]
Например, W означает, что либо аденин, либо тимин могут находиться в этом положении без нарушения функциональности последовательности.
| Символ [2] | Значение/происхождение | Возможные базы | Дополнить | ||||
|---|---|---|---|---|---|---|---|
| А | Денин | А | 1 | Т (или У) | |||
| С | Цитозин | С | Г | ||||
| Г | Гуанин | Г | С | ||||
| Т | Т химин | Т | А | ||||
| В | Ты расовый | В | А | ||||
| В | Слабый | А | Т | 2 | В | ||
| С | Сильный | С | Г | С | |||
| М | Мино | А | С | К | |||
| К | К это | Г | Т | М | |||
| Р | Пу Рин | А | Г | И | |||
| И | p Y римидин | С | Т | Р | |||
| Б | не А ( Б идет после А) | С | Г | Т | 3 | V | |
| Д | не C ( D идет после C) | А | Г | Т | ЧАС | ||
| ЧАС | не G ( H идет после G) | А | С | Т | Д | ||
| V | не T ( V идет после T и U) | А | С | Г | Б | ||
| Н | любой нуклеотид (не пробел) | А | С | Г | Т | 4 | Н |
| С | ноль | 0 | С | ||||
Эти символы также действительны для РНК, за исключением того, что U (урацил) заменяет Т (тимин). [1]
Помимо аденина (А), цитозина (С), гуанина (G), тимина (Т) и урацила (U), ДНК и РНК также содержат основания, модифицированные после образования цепи нуклеиновой кислоты. В ДНК наиболее распространенным модифицированным основанием является 5-метилцитидин (m5C). В РНК имеется множество модифицированных оснований, в том числе псевдоуридин (Ψ), дигидроуридин (D), инозин (I), риботимидин (rT) и 7-метилгуанозин (m7G). [3] [4] Гипоксантин и ксантин — два из многих оснований, созданных в результате присутствия мутагена , причем оба они являются результатом дезаминирования (замены аминогруппы карбонильной группой). Гипоксантин производится из аденина , а ксантин — из гуанина . [5] Аналогичным образом дезаминирование цитозина приводит к образованию урацила .
- Пример сравнения и определения % разницы между двумя нуклеотидными последовательностями
- AA T CC GC ТЕГ
- AA A CC CT ТЕГ
Учитывая две 10-нуклеотидные последовательности, выровняйте их и сравните различия между ними. Рассчитайте процентную разницу, разделив количество различий между основаниями ДНК на общее количество нуклеотидов. В этом случае имеется три различия в последовательности из 10 нуклеотидов. Таким образом, разница составляет 30%.
значение Биологическое
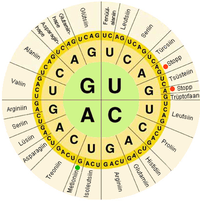
В биологических системах нуклеиновые кислоты содержат информацию, которая используется живой клеткой для построения специфических белков . Последовательность нуклеиновых оснований на цепи нуклеиновой кислоты транслируется клеточным механизмом в последовательность аминокислот, составляющих белковую цепь. Каждая группа из трех оснований, называемая кодоном , соответствует одной аминокислоте, и существует определенный генетический код , согласно которому каждая возможная комбинация трех оснований соответствует определенной аминокислоте.
Центральная догма молекулярной биологии описывает механизм, с помощью которого белки конструируются с использованием информации, содержащейся в нуклеиновых кислотах. ДНК транскрибируется , в молекулы мРНК , которые попадают в рибосому где мРНК используется в качестве матрицы для построения белковой цепи. Поскольку нуклеиновые кислоты могут связываться с молекулами с комплементарными последовательностями, существует различие между « смысловыми » последовательностями, кодирующими белки, и комплементарной «антисмысловой» последовательностью, которая сама по себе нефункциональна, но может связываться со смысловой цепью.
Определение последовательности [ править ]
Секвенирование ДНК — это процесс определения нуклеотидной последовательности данного фрагмента ДНК . Последовательность ДНК живого существа кодирует необходимую информацию для выживания и размножения живого существа. Поэтому определение последовательности полезно в фундаментальных исследованиях того, почему и как живут организмы, а также в прикладных предметах. Из-за важности ДНК для живых существ знание последовательности ДНК может быть полезно практически в любом биологическом исследовании . Например, в медицине его можно использовать для выявления, диагностики и потенциальной разработки методов лечения генетических заболеваний . Аналогичным образом, исследования патогенов могут привести к разработке методов лечения инфекционных заболеваний. Биотехнология – это развивающаяся дисциплина, имеющая потенциал для создания множества полезных продуктов и услуг.
РНК не секвенируется напрямую. Вместо этого она копируется в ДНК с помощью обратной транскриптазы , и эта ДНК затем секвенируется.
Современные методы секвенирования основаны на дискриминационной способности ДНК-полимераз и, следовательно, могут различать только четыре основания. Инозин (созданный из аденозина при редактировании РНК ) читается как G, а 5-метилцитозин (созданный из цитозина путем метилирования ДНК ) читается как C. При нынешних технологиях сложно секвенировать небольшие количества ДНК. поскольку сигнал слишком слаб для измерения. Эту проблему можно преодолеть с помощью амплификации полимеразной цепной реакции (ПЦР).
Цифровое представление [ править ]

После того как последовательность нуклеиновой кислоты получена из организма, она сохраняется в silico в цифровом формате. Цифровые генетические последовательности могут храниться в базах данных последовательностей , анализироваться (см. «Анализ последовательностей» ниже), изменяться в цифровом виде и использоваться в качестве шаблонов для создания новой фактической ДНК с использованием искусственного синтеза генов .
Анализ последовательности [ править ]
Цифровые генетические последовательности можно анализировать с помощью инструментов биоинформатики , чтобы попытаться определить их функцию.
Генетическое тестирование [ править ]
организма ДНК в геноме можно анализировать для диагностики уязвимости к наследственным заболеваниям человека , а также использовать для определения отцовства ребенка (генетического отца) или происхождения . Обычно каждый человек несет в себе две вариации каждого гена : одну от матери, другую от отца. генов . Считается, что геном человека содержит около 20 000–25 000 Помимо изучения хромосом до уровня отдельных генов, генетическое тестирование в более широком смысле включает в себя биохимические тесты на возможное наличие генетических заболеваний или мутантных форм генов, связанных с повышенным риском развития генетических нарушений.
Генетическое тестирование выявляет изменения в хромосомах, генах или белках. [6] Обычно тестирование используется для обнаружения изменений, связанных с наследственными заболеваниями. Результаты генетического теста могут подтвердить или исключить предполагаемое генетическое заболевание или помочь определить вероятность развития или передачи генетического заболевания у человека. В настоящее время используется несколько сотен генетических тестов, и еще больше разрабатывается. [7] [8]
Выравнивание последовательности [ править ]
В биоинформатике выравнивание последовательностей — это способ упорядочения последовательностей ДНК , РНК или белка для выявления областей сходства, которое может быть связано с функциональными, структурными или эволюционными отношениями между последовательностями. [9] Если две последовательности в выравнивании имеют общего предка, несоответствия можно интерпретировать как точечные мутации , а пробелы — как мутации вставки или делеции ( индели ), возникшие в одной или обеих линиях с момента их расхождения друг от друга. При выравнивании последовательностей белков степень сходства между аминокислотами , занимающими определенное положение в последовательности, можно интерпретировать как приблизительную меру того, насколько консервативен конкретный участок или мотив последовательности среди линий. Отсутствие замен или наличие только очень консервативных замен (то есть замены аминокислот, боковые цепи которых имеют сходные биохимические свойства) в определенном участке последовательности предполагают [10] что этот регион имеет структурное или функциональное значение. основания ДНК и РНК Хотя нуклеотидные более похожи друг на друга, чем аминокислоты, консервативность пар оснований может указывать на сходную функциональную или структурную роль. [11]
Вычислительная филогенетика широко использует выравнивание последовательностей при построении и интерпретации филогенетических деревьев , которые используются для классификации эволюционных отношений между гомологичными генами, представленными в геномах дивергентных видов. Степень различия последовательностей в наборе запросов качественно связана с эволюционным расстоянием последовательностей друг от друга. Грубо говоря, высокая идентичность последовательностей предполагает, что рассматриваемые последовательности имеют сравнительно молодого самого недавнего общего предка , тогда как низкая идентичность предполагает, что расхождение более древнее. Это приближение, которое отражает гипотезу « молекулярных часов », согласно которой примерно постоянная скорость эволюционных изменений может быть использована для экстраполяции времени, прошедшего с тех пор, как два гена впервые разошлись (то есть время слияния ), предполагает, что эффекты мутации отбора и константа во всех линиях последовательности. Следовательно, он не учитывает возможные различия между организмами или видами в скорости Репарация ДНК или возможная функциональная консервация определенных участков последовательности. (В случае нуклеотидных последовательностей гипотеза молекулярных часов в своей самой базовой форме также не учитывает разницу в скорости принятия между «молчащими» мутациями , которые не меняют значения данного кодона другой аминокислоты , и другими мутациями, которые приводят к включению в структуру ). белок.) Более статистически точные методы позволяют варьировать скорость эволюции на каждой ветви филогенетического дерева, что позволяет лучше оценить время слияния генов.
Последовательность мотивов [ править ]
Часто первичная структура кодирует мотивы, имеющие функциональное значение. Некоторые примеры мотивов последовательности: C/D [12] и коробки H/ACA [13] мякРНК , , сайт связывания Sm обнаруженный в сплайсосомальных РНК, таких как U1 , U2 , U4 , U5 , U6 , U12 и U3 , последовательность Шайна-Дальгарно , [14] Козака консенсусная последовательность [15] и терминатор РНК-полимеразы III . [16]
Энтропия последовательности [ править ]
В биоинформатике энтропия последовательности, также известная как сложность последовательности или информационный профиль, [17] представляет собой числовую последовательность, обеспечивающую количественную меру локальной сложности последовательности ДНК независимо от направления обработки. Манипуляции с информационными профилями позволяют анализировать последовательности с использованием методов без выравнивания, таких как, например, обнаружение мотивов и перегруппировок. [17] [18] [19]
См. также [ править ]
- Генная структура
- Определение структуры нуклеиновой кислоты
- Четвертичная система счисления
- Однонуклеотидный полиморфизм (SNP)
Ссылки [ править ]
- ^ Jump up to: Перейти обратно: а б «Номенклатура не полностью определенных оснований в последовательностях нуклеиновых кислот. Рекомендации 1984 года. Номенклатурный комитет Международного союза биохимии (NC-IUB)» . Труды Национальной академии наук . 83 (1): 4–8. 1986. дои : 10.1073/pnas.83.1.4 . ISSN 0027-8424 . ПМК 322779 . ПМИД 2417239 .
- ^ Номенклатурный комитет Международного биохимического союза (NC-IUB) (1984). «Номенклатура не полностью определенных оснований в последовательностях нуклеиновых кислот» . Проверено 4 февраля 2008 г.
- ^ «БИОЛ2060: Перевод» . mun.ca.
- ^ "Исследовать" . uw.edu.pl.
- ^ Нгуен, Т; Брансон, Д; Креспи, CL; Пенман, BW; Вишнок, Дж.С.; Танненбаум, СР (апрель 1992 г.). «Повреждение и мутация ДНК в клетках человека, подвергшихся воздействию оксида азота in vitro» . Proc Natl Acad Sci США . 89 (7): 3030–034. Бибкод : 1992PNAS...89.3030N . дои : 10.1073/pnas.89.7.3030 . ПМК 48797 . ПМИД 1557408 .
- ^ «Что такое генетическое тестирование?» . Домашний справочник по генетике . 16 марта 2015 года. Архивировано из оригинала 29 мая 2006 года . Проверено 19 мая 2010 г.
- ^ «Генетическое тестирование» . nih.gov .
- ^ «Определения генетического тестирования» . Определения генетического тестирования (Хорхе Секейрос и Барбара Гимарайнш) . Проект сети передового опыта EuroGentest. 11 сентября 2008 г. Архивировано из оригинала 4 февраля 2009 года . Проверено 10 августа 2008 г.
- ^ Крепление ДМ. (2004). Биоинформатика: анализ последовательностей и генома (2-е изд.). Лабораторное издательство Колд-Спринг-Харбор: Колд-Спринг-Харбор, Нью-Йорк. ISBN 0-87969-608-7 .
- ^ Нг, ПК; Хеникофф, С. (2001). «Прогнозирование вредных аминокислотных замен» . Геномные исследования . 11 (5): 863–74. дои : 10.1101/гр.176601 . ПМК 311071 . ПМИД 11337480 .
- ^ Вицани, Г. (2016). «Важные шаги к жизни: от химических реакций к кодированию с использованием агентов» . Биосистемы . 140 : 49–57. Бибкод : 2016BiSys.140...49Вт . doi : 10.1016/j.biosystems.2015.12.007 . ПМИД 26723230 . S2CID 30962295 .
- ^ Самарский Д.А.; Фурнье МЖ; Певица Р.Х.; Бертран Э (1998). «Мотив C/D-бокса мякРНК управляет ядрышковым нацеливанием, а также связывает синтез и локализацию мякРНК» . Журнал ЭМБО . 17 (13): 3747–57. дои : 10.1093/emboj/17.13.3747 . ПМК 1170710 . ПМИД 9649444 .
- ^ Гано, Филипп; Кайзерг-Феррер, Мишель; Поцелуй, Тамаш (1 апреля 1997 г.). «Семейство малых ядрышковых РНК бокса ACA определяется эволюционно консервативной вторичной структурой и повсеместными элементами последовательности, необходимыми для накопления РНК» . Гены и развитие . 11 (7): 941–56. дои : 10.1101/gad.11.7.941 . ПМИД 9106664 .
- ^ Шайн Дж., Далгарно Л. (1975). «Определитель цистронной специфичности бактериальных рибосом». Природа . 254 (5495): 34–38. Бибкод : 1975Natur.254...34S . дои : 10.1038/254034a0 . ПМИД 803646 . S2CID 4162567 .
- ^ Козак М. (октябрь 1987 г.). «Анализ 5'-некодирующих последовательностей 699 информационных РНК позвоночных» . Нуклеиновые кислоты Рез . 15 (20): 8125–48. дои : 10.1093/нар/15.20.8125 . ПМК 306349 . ПМИД 3313277 .
- ^ Богенхаген Д.Ф., Браун Д.Д. (1981). «Нуклеотидные последовательности в ДНК Xenopus 5S, необходимые для терминации транскрипции». Клетка . 24 (1): 261–70. дои : 10.1016/0092-8674(81)90522-5 . ПМИД 6263489 . S2CID 9982829 .
- ^ Jump up to: Перейти обратно: а б Пиньо, А; Гарсия, С; Пратас, Д; Феррейра, П. (21 ноября 2013 г.). «Краткий обзор последовательностей ДНК» . ПЛОС ОДИН . 8 (11): е79922. Бибкод : 2013PLoSO...879922P . дои : 10.1371/journal.pone.0079922 . ПМЦ 3836782 . ПМИД 24278218 .
- ^ Пратас, Д; Сильва, Р; Пиньо, А; Феррейра, П. (18 мая 2015 г.). «Метод без выравнивания для поиска и визуализации перестроек между парами последовательностей ДНК» . Научные отчеты . 5 : 10203. Бибкод : 2015NatSR...510203P . дои : 10.1038/srep10203 . ПМЦ 4434998 . ПМИД 25984837 .
- ^ Троянская, О; Арбелл, О; Корен, Ю; Ландау, Г; Большой, А (2002). «Профили сложности последовательностей геномных последовательностей прокариот: быстрый алгоритм расчета лингвистической сложности» . Биоинформатика . 18 (5): 679–88. дои : 10.1093/биоинформатика/18.5.679 . ПМИД 12050064 .
Внешние ссылки [ править ]
| Базы данных органов управления : Национальные |
|---|