Дизайн лекарств
Разработка лекарств , часто называемая рациональным дизайном лекарств или просто рациональным дизайном , представляет собой изобретательский процесс поиска новых лекарств, основанный на знании биологической мишени . [1] Препарат чаще всего представляет собой небольшую органическую молекулу , которая активирует или ингибирует функцию биомолекулы, такой как белок , что, в свою очередь, приводит к терапевтическому эффекту для пациента . В самом простом смысле дизайн лекарств включает в себя создание молекул, которые по форме и заряду комплементарны биомолекулярной мишени, с которой они взаимодействуют и, следовательно, будут связываться с ней. Разработка лекарств часто, но не обязательно, опирается на методы компьютерного моделирования . [2] Этот тип моделирования иногда называют компьютерным дизайном лекарств . Наконец, разработка лекарств, основанная на знании трехмерной структуры биомолекулярной мишени, известна как разработка лекарств на основе структуры . [2] Помимо малых молекул, биофармацевтические препараты, включая пептиды [3] [4] и особенно терапевтические антитела становятся все более важным классом лекарств, и также были разработаны вычислительные методы для улучшения аффинности, селективности и стабильности этих терапевтических средств на основе белков. [5]
Определение
[ редактировать ]Фраза «дизайн лекарства» аналогична дизайну лиганда (т.е. дизайну молекулы, которая будет прочно связываться со своей мишенью). [6] Хотя методы проектирования для прогнозирования аффинности связывания достаточно успешны, существует множество других свойств, таких как биодоступность , период метаболического полувыведения и побочные эффекты , которые сначала необходимо оптимизировать, прежде чем лиганд сможет стать безопасным и эффективным лекарственным средством. Эти другие характеристики часто трудно предсказать с помощью методов рационального проектирования.
Из-за высоких темпов отсева, особенно на клинических этапах разработки лекарств , на ранних этапах процесса разработки лекарств больше внимания уделяется выбору лекарств-кандидатов, чьи физико-химические свойства, по прогнозам, приведут к меньшему количеству осложнений во время разработки и, следовательно, с большей вероятностью приведут к одобрению препарата. , продаваемый препарат. [7] Кроме того, эксперименты in vitro, дополненные вычислительными методами, все чаще используются на ранних этапах открытия лекарств для выбора соединений с более благоприятными ADME (абсорбция, распределение, метаболизм и выведение) и токсикологическими профилями. [8]
Цели по наркотикам
[ редактировать ]Биомолекулярная мишень (чаще всего белок или нуклеиновая кислота ) представляет собой ключевую молекулу, участвующую в определенном метаболическом или сигнальном пути, который связан с конкретным болезненным состоянием или патологией или с инфекционностью или выживанием микробного патогена . Потенциальные мишени для лекарств не обязательно вызывают заболевание, но по определению должны модифицировать заболевание. [9] В некоторых случаях будут разработаны небольшие молекулы для усиления или ингибирования целевой функции на пути изменения конкретного заболевания. Малые молекулы (например , агонисты , антагонисты , обратные агонисты или модуляторы ферментов рецепторов; активаторы или ингибиторы ; открыватели или блокаторы ионных каналов ) [10] будут разработаны так, чтобы они были комплементарны сайту связывания мишени. [11] Малые молекулы (лекарства) могут быть разработаны так, чтобы не воздействовать на другие важные молекулы, «нецелевые» (часто называемые антимишенями ), поскольку взаимодействие лекарства с молекулами, не попадающими в цель, может привести к нежелательным побочным эффектам . [12] Из-за сходства сайтов связывания близкородственные мишени, идентифицированные посредством гомологии последовательностей, имеют самую высокую вероятность перекрестной реактивности и, следовательно, самый высокий потенциал побочных эффектов.
Чаще всего лекарства представляют собой органические небольшие молекулы, производимые посредством химического синтеза, но лекарства на основе биополимеров (также известные как биофармацевтические препараты ), производимые посредством биологических процессов, становятся все более распространенными. [13] Кроме того, мРНК могут иметь терапевтическое применение. на основе подавления генов технологии [14] Например, наномедицины на основе мРНК могут упростить и ускорить процесс разработки лекарств, обеспечивая временную и локализованную экспрессию иммуностимулирующих молекул. [15] Транскрибируемая in vitro (IVT) мРНК позволяет доставлять ее в различные доступные типы клеток через кровь или альтернативными путями. Использование мРНК IVT служит для передачи специфической генетической информации в клетки человека с основной целью предотвращения или изменения конкретного заболевания. [16]
Открытие лекарств
[ редактировать ]Открытие фенотипического лекарства
[ редактировать ]Открытие фенотипических лекарств — это традиционный метод открытия лекарств, также известный как передовая фармакология или классическая фармакология. Он использует процесс фенотипического скрининга коллекций синтетических малых молекул, натуральных продуктов или экстрактов в химических библиотеках, чтобы точно определить вещества, обладающие полезным терапевтическим эффектом. Этот метод заключается в том, чтобы сначала обнаружить функциональную активность лекарств in vivo или in vitro (таких как экстракты лекарств или натуральные продукты), а затем выполнить идентификацию цели. Фенотипическое открытие использует практический и независимый от цели подход для привлечения первоначальных потенциальных клиентов с целью обнаружения фармакологически активных соединений и терапевтических средств, которые действуют через новые лекарственные механизмы. [17] Этот метод позволяет исследовать фенотипы заболеваний и находить потенциальные методы лечения состояний неизвестного, сложного или многофакторного происхождения, когда понимание молекулярных мишеней недостаточно для эффективного вмешательства. [18]
Рациональное открытие лекарств
[ редактировать ]Рациональный дизайн лекарств (также называемый обратной фармакологией ) начинается с гипотезы о том, что модуляция конкретной биологической мишени может иметь терапевтическую ценность. Чтобы выбрать биомолекулу в качестве мишени для лекарства, необходимы две важные части информации. Во-первых, это свидетельство того, что модуляция мишени будет модифицировать заболевание. Эти знания могут быть получены, например, из исследований связи заболеваний, которые показывают связь между мутациями в биологической мишени и определенными болезненными состояниями. [19] Во-вторых, мишень способна связываться с небольшой молекулой и что ее активность может модулироваться небольшой молекулой. [20]
Как только подходящая мишень идентифицирована, ее обычно клонируют , производят и очищают . Очищенный белок затем используется для проведения скринингового анализа . Кроме того, может быть определена трехмерная структура мишени.
Поиск малых молекул, которые связываются с мишенью, начинается с скрининга библиотек потенциальных лекарственных соединений. Это можно сделать с помощью скринингового анализа («мокрый скрининг»). Кроме того, если доступна структура мишени, можно провести виртуальный скрининг потенциальных лекарств. В идеале соединения-кандидаты в лекарственные средства должны быть « подобны лекарственным средствам », то есть они должны обладать свойствами, которые, как ожидается, приведут к биодоступности при пероральном приеме , адекватной химической и метаболической стабильности и минимальным токсическим эффектам. [21] Для оценки сходства лекарств доступно несколько методов, таких как «Правило пяти» Липински , а также ряд методов оценки, таких как липофильная эффективность . [22] В научной литературе также предложено несколько методов прогнозирования метаболизма лекарств. [23]
Из-за большого количества свойств лекарств, которые необходимо одновременно оптимизировать в процессе проектирования, многокритериальной оптимизации . иногда используются методы [24] Наконец, из-за ограничений существующих методов прогнозирования активности разработка лекарств по-прежнему во многом зависит от счастливой случайности. [25] и ограниченная рациональность . [26]
Компьютерный дизайн лекарств
[ редактировать ]Самая фундаментальная цель разработки лекарств — предсказать, будет ли данная молекула связываться с мишенью, и если да, то насколько сильно. Молекулярная механика или молекулярная динамика чаще всего используется для оценки силы межмолекулярного взаимодействия между небольшой молекулой и ее биологической мишенью. Эти методы также используются для прогнозирования конформации небольшой молекулы и моделирования конформационных изменений мишени, которые могут произойти, когда небольшая молекула связывается с ней. [3] [4] Полуэмпирические или методы квантовой химии ab initio теория функционала плотности часто используются для получения оптимизированных параметров для расчетов молекулярной механики, а также для оценки электронных свойств (электростатический потенциал, поляризуемость и т. д.) кандидата на лекарство, которое будет влияют на аффинность связывания. [27]
Методы молекулярной механики также можно использовать для полуколичественного прогнозирования аффинности связывания. основанную на знаниях функцию оценки, Кроме того, для оценки аффинности связывания можно использовать . Эти методы используют линейную регрессию , машинное обучение , нейронные сети или другие статистические методы для получения прогнозирующих уравнений сродства связывания путем подгонки экспериментального сродства к полученным с помощью вычислений энергиям взаимодействия между небольшой молекулой и мишенью. [28] [29]
В идеале вычислительный метод сможет предсказать сродство до того, как соединение будет синтезировано, и, следовательно, теоретически необходимо синтезировать только одно соединение, что экономит огромное время и деньги. Реальность такова, что существующие вычислительные методы несовершенны и в лучшем случае дают только качественно точные оценки сродства. На практике требуется несколько итераций проектирования, синтеза и испытаний, прежде чем будет найден оптимальный препарат. Вычислительные методы ускорили открытия за счет сокращения количества необходимых итераций и часто позволяли создавать новые структуры. [30] [31]
Компьютерный дизайн лекарств может использоваться на любом из следующих этапов открытия лекарств:
- идентификация попаданий с использованием виртуального скрининга (дизайн на основе структуры или лиганда)
- при попадании к лиду оптимизация сродства и селективности (структурно-ориентированный дизайн, QSAR и т. д.)
- провести оптимизацию других фармацевтических свойств при сохранении аффинности

Чтобы преодолеть недостаточное предсказание аффинности связывания, рассчитанной с помощью недавних оценочных функций, для анализа используется взаимодействие белок-лиганд и информация о трехмерной структуре соединения. Для разработки лекарств на основе структуры было разработано несколько анализов после скрининга, ориентированных на взаимодействие белка и лиганда, для улучшения обогащения и эффективного выявления потенциальных кандидатов:
- Консенсусная оценка [32] [33]
- Отбор кандидатов путем голосования по нескольким оценочным функциям
- Может потерять связь между структурной информацией белок-лиганд и критерием оценки.
- Кластерный анализ [34] [35]
- Представляйте и группируйте кандидатов в соответствии с трехмерной информацией о белке-лиганде.
- Требуется значимое представление взаимодействий белок-лиганд.
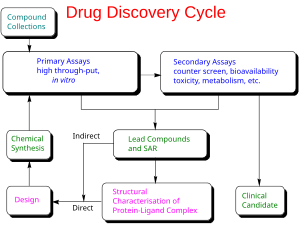
Типы
[ редактировать ]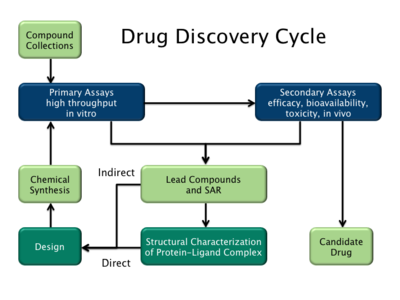
Существует два основных типа дизайна лекарств. Первый называется разработкой лекарств на основе лигандов , а второй — разработкой лекарств на основе структуры. [2]
на основе лигандов
[ редактировать ]Разработка лекарств на основе лигандов (или непрямая разработка лекарств ) опирается на знание других молекул, которые связываются с интересующей биологической мишенью. Эти другие молекулы могут быть использованы для создания модели фармакофора , которая определяет минимально необходимые структурные характеристики, которыми должна обладать молекула для связывания с мишенью. [36] Модель биологической мишени может быть построена на основе знаний о том, что с ней связывается, а эта модель, в свою очередь, может использоваться для создания новых молекулярных объектов, которые взаимодействуют с мишенью. В качестве альтернативы можно получить количественную зависимость структура-активность (QSAR), в которой существует корреляция между расчетными свойствами молекул и их экспериментально определенной биологической активностью . Эти отношения QSAR, в свою очередь, можно использовать для прогнозирования активности новых аналогов. [37]
Структурно-ориентированный
[ редактировать ]Структурно-ориентированный дизайн лекарств (или прямой дизайн лекарств ) основан на знании трехмерной структуры биологической мишени, полученной с помощью таких методов, как рентгеновская кристаллография или ЯМР-спектроскопия . [38] Если экспериментальная структура мишени недоступна, возможно создать модель гомологии мишени на основе экспериментальной структуры родственного белка. Используя структуру биологической мишени, лекарства-кандидаты, которые, по прогнозам, будут связываться с с высоким сродством и селективностью мишенью , могут быть разработаны с использованием интерактивной графики и интуиции фармацевта-медика . Альтернативно, для предложения новых кандидатов на лекарственные средства можно использовать различные автоматизированные вычислительные процедуры. [39]
Современные методы разработки лекарств на основе структуры можно условно разделить на три основные категории. [40] Первый метод — это идентификация новых лигандов для данного рецептора путем поиска в больших базах данных трехмерных структур малых молекул с целью поиска тех, которые подходят для связывающего кармана рецептора, с использованием программ быстрой аппроксимационной стыковки . Этот метод известен как виртуальный скрининг .
Вторая категория — это создание новых лигандов de novo. В этом методе молекулы-лиганды создаются в пределах связывающего кармана путем поэтапной сборки небольших частей. Эти кусочки могут быть как отдельными атомами, так и фрагментами молекул. Ключевое преимущество такого метода заключается в том, что можно предложить новые структуры, которых нет ни в одной базе данных. [41] [42] [43] Третий метод — оптимизация известных лигандов путем оценки предлагаемых аналогов внутри полости связывания. [40]
Идентификация сайта привязки
[ редактировать ]Идентификация сайта связывания является первым шагом в структурном проектировании. [20] [44] Если структура мишени или достаточно близкого гомолога определяется в присутствии связанного лиганда, то лиганд должен быть виден в структуре, и в этом случае расположение сайта связывания тривиально. Однако могут существовать незанятые аллостерические сайты связывания , которые могут представлять интерес. Более того, возможно, что доступны только структуры апопротеина (белка без лиганда), и надежная идентификация незанятых сайтов, которые могут связывать лиганды с высоким сродством, является нетривиальной задачей. Короче говоря, идентификация сайта связывания обычно основана на идентификации вогнутых поверхностей на белке, на которых могут размещаться молекулы размера лекарственного средства, которые также имеют соответствующие «горячие точки» ( гидрофобные поверхности, сайты водородных связей и т. д.), которые управляют связыванием лиганда. [20] [44]
Функции оценки
[ редактировать ]Структурно-ориентированный дизайн лекарств пытается использовать структуру белков в качестве основы для разработки новых лигандов, применяя принципы молекулярного распознавания . Обычно желательно селективное связывание с высоким сродством к мишени, поскольку оно приводит к созданию более эффективных лекарств с меньшим количеством побочных эффектов. Таким образом, одним из наиболее важных принципов проектирования или получения потенциальных новых лигандов является предсказание сродства связывания определенного лиганда с его мишенью (и известными антимишенями ) и использование предсказанного сродства в качестве критерия отбора. [45]
Одна из первых универсальных эмпирических оценочных функций для описания энергии связывания лигандов с рецепторами была разработана Бёмом. [46] [47] Эта эмпирическая оценочная функция имела вид:

где:
- ΔG 0 – эмпирически полученное смещение, которое частично соответствует общей потере трансляционной и вращательной энтропии лиганда при связывании.
- ΔG hb – вклад водородной связи
- ΔG ионный - вклад ионных взаимодействий
- ΔG Lip – вклад липофильных взаимодействий, где |A липо | - площадь поверхности липофильного контакта между лигандом и рецептором.
- ΔG rot - штраф за энтропию из-за замораживания вращающегося компонента в лигандной связи при связывании.
Более общее термодинамическое «главное» уравнение выглядит следующим образом: [48]
![{\displaystyle {\begin{array}{lll}\Delta G_{\text{bind}}=-RT\ln K_{\text{d}}\\[1.3ex]K_{\text{d}}= {\dfrac {[{\text{Лиганд}}][{\text{Рецептор}}]}{[{\text{Комплекс}}]}}\\[1.3ex]\Delta G_{\text{bind} }=\Delta G_{\text{десольвация}}+\Delta G_{\text{движение}}+\Delta G_{\text{configuration}}+\Delta G_{\text{interaction}}\end{array} }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ba49ddd9dec7415d129787213744ca1afcd2d021)
где:
- десольвация - энтальпийная плата за удаление лиганда из растворителя.
- движение - энтропийный штраф за уменьшение степеней свободы, когда лиганд связывается со своим рецептором.
- конфигурация - энергия конформационной деформации, необходимая для перевода лиганда в «активную» конформацию.
- взаимодействие - энтальпийный выигрыш за «разрешение» лиганда с его рецептором.
Основная идея состоит в том, что общую свободную энергию связывания можно разложить на независимые компоненты, которые, как известно, важны для процесса связывания. Каждый компонент отражает определенный вид изменения свободной энергии во время процесса связывания лиганда с его целевым рецептором. Главное уравнение представляет собой линейную комбинацию этих компонентов. По уравнению свободной энергии Гиббса построена связь между константой равновесия диссоциации K d и компонентами свободной энергии.
Для оценки каждого из компонентов основного уравнения используются различные вычислительные методы. Например, изменение площади полярной поверхности при связывании лиганда можно использовать для оценки энергии десольватации. Количество вращающихся связей, замороженных при связывании лиганда, пропорционально члену движения. Конфигурационную энергию или энергию деформации можно оценить с помощью расчетов молекулярной механики . Наконец, энергию взаимодействия можно оценить с помощью таких методов, как изменение неполярной поверхности, статистически полученные потенциалы средней силы , количество образующихся водородных связей и т. д. На практике компоненты основного уравнения подгоняются к экспериментальным данным с использованием нескольких линейная регрессия. Это можно сделать с помощью разнообразного обучающего набора, включающего множество типов лигандов и рецепторов для создания менее точной, но более общей «глобальной» модели, или более ограниченного набора лигандов и рецепторов для создания более точной, но менее общей «локальной» модели. [49]
Примеры
[ редактировать ]Конкретный пример рационального дизайна лекарств включает использование трехмерной информации о биомолекулах, полученной с помощью таких методов, как рентгеновская кристаллография и ЯМР-спектроскопия. Компьютерный дизайн лекарств, в частности, становится гораздо более простым, когда имеется структура целевого белка с высоким разрешением, связанного с мощным лигандом. Такой подход к открытию лекарств иногда называют структурно-ориентированным дизайном лекарств. Первым однозначным примером применения структурного дизайна лекарств, приведшего к одобрению препарата, является ингибитор карбоангидразы дорзоламид , который был одобрен в 1995 году. [50] [51]
Еще одним примером рационального дизайна лекарств является иматиниб , ингибитор тирозинкиназы , разработанный специально для слитого белка bcr-abl , который характерен для филадельфийской хромосомой с лейкозов ( хронический миелогенный лейкоз и иногда острый лимфоцитарный лейкоз ). Иматиниб существенно отличается от предыдущих лекарств от рака , поскольку большинство агентов химиотерапии просто нацелены на быстро делящиеся клетки, не различая раковые клетки и другие ткани. [52]
Дополнительные примеры включают в себя:
- Многие из атипичных нейролептиков
- Циметидин , прототипный антагонист H 2 -рецепторов , из которого произошли более поздние представители этого класса.
- Селективные ЦОГ-2 ингибиторы НПВП
- Энфувиртид — пептидный ингибитор проникновения ВИЧ.
- Небензодиазепины, такие как золпидем и зопиклон.
- Ралтегравир – интегразы ВИЧ. ингибитор [53]
- СИОЗС (селективные ингибиторы обратного захвата серотонина), класс антидепрессантов.
- Занамивир , противовирусный препарат
Проверка на наркотики
[ редактировать ]Типы скрининга на наркотики включают фенотипический скрининг , высокопроизводительный скрининг и виртуальный скрининг . Фенотипический скрининг характеризуется процессом скрининга лекарств с использованием моделей клеточных или животных заболеваний для выявления соединений, которые изменяют фенотип и оказывают благоприятное воздействие на заболевание. [54] [55] Новые технологии высокопроизводительного скрининга существенно повышают скорость обработки и уменьшают необходимый объем обнаружения. [56] Виртуальный скрининг выполняется с помощью компьютера, что позволяет проверять большое количество молекул за короткий цикл и с низкими затратами. Виртуальный скрининг использует ряд вычислительных методов, которые позволяют химикам сокращать обширные виртуальные библиотеки до более управляемых размеров. [57]
Тематические исследования
[ редактировать ]- Антагонисты 5-НТ3
- Агонисты рецепторов ацетилхолина
- Антагонисты рецепторов ангиотензина
- Ингибиторы тирозинкиназы Bcr-Abl
- Антагонисты каннабиноидных рецепторов
- Антагонисты рецептора CCR5
- Ингибиторы циклооксигеназы 2
- Ингибиторы дипептидилпептидазы-4
- Ингибиторы протеазы ВИЧ
- Антагонисты рецепторов NK1
- Ненуклеозидные ингибиторы обратной транскриптазы
- Нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы
- Ингибиторы ФДЭ5
- Ингибиторы протонной помпы
- Ингибиторы ренина
- Триптаны
- Антагонисты TRPV1
- ингибиторы c-Met
Критика
[ редактировать ]Утверждалось, что очень жесткий и целенаправленный характер рационального дизайна лекарств подавляет случайность при открытии лекарств. [58]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Мадсен У., Крогсгаард-Ларсен П., Лильефорс Т. (2002). Учебник по разработке и открытию лекарств . Вашингтон, округ Колумбия: Тейлор и Фрэнсис. ISBN 978-0-415-28288-8 .
- ^ Jump up to: а б с Рейнольдс Ч., Мерц К.М., Ринге Д., ред. (2010). Разработка лекарств: подходы на основе структуры и лигандов (1-е изд.). Кембридж, Великобритания: Издательство Кембриджского университета. ISBN 978-0521887236 .
- ^ Jump up to: а б Фосгерау К., Хоффманн Т. (январь 2015 г.). «Пептидная терапия: современное состояние и будущие направления» . Открытие наркотиков сегодня . 20 (1): 122–128. дои : 10.1016/j.drudis.2014.10.003 . ПМИД 25450771 .
- ^ Jump up to: а б Циемни М., Курчински М., Камель К., Колински А., Алам Н., Шулер-Фурман О., Кмиецик С. (август 2018 г.). «Белко-пептидный докинг: возможности и проблемы» . Открытие наркотиков сегодня . 23 (8): 1530–1537. дои : 10.1016/j.drudis.2018.05.006 . ПМИД 29733895 .
- ^ Шираи Х., Прадес С., Вита Р., Маркатили П., Попович Б., Сюй Дж. и др. (ноябрь 2014 г.). «Информатика антител для открытия лекарств». Biochimica et Biophysical Acta (BBA) - Белки и протеомика . 1844 (11): 2002–2015. дои : 10.1016/j.bbapap.2014.07.006 . ПМИД 25110827 .
- ^ Толленаэре Дж. П. (апрель 1996 г.). «Роль структурного дизайна лигандов и молекулярного моделирования в открытии лекарств». Аптечный мир и наука . 18 (2): 56–62. дои : 10.1007/BF00579706 . ПМИД 8739258 . S2CID 21550508 .
- ^ Уоринг М.Дж., Эрроусмит Дж., Лич А.Р., Лисон П.Д., Мандрелл С., Оуэн Р.М. и др. (июль 2015 г.). «Анализ истощения кандидатов на лекарства из четырех крупных фармацевтических компаний». Обзоры природы. Открытие наркотиков . 14 (7): 475–486. дои : 10.1038/nrd4609 . ПМИД 26091267 . S2CID 25292436 .
- ^ Ю Х, Адедоин А (сентябрь 2003 г.). «ADME-Tox в открытии лекарств: интеграция экспериментальных и вычислительных технологий». Открытие наркотиков сегодня . 8 (18): 852–861. дои : 10.1016/S1359-6446(03)02828-9 . ПМИД 12963322 .
- ^ Диксон С.Дж., Стоквелл Б.Р. (декабрь 2009 г.). «Идентификация лекарственных генных продуктов, модифицирующих болезнь» . Современное мнение в области химической биологии . 13 (5–6): 549–555. дои : 10.1016/j.cbpa.2009.08.003 . ПМК 2787993 . ПМИД 19740696 .
- ^ Имминг П., Синнинг С., Мейер А. (октябрь 2006 г.). «Наркотики, их мишени, а также характер и количество мишеней для наркотиков». Обзоры природы. Открытие наркотиков . 5 (10): 821–834. дои : 10.1038/nrd2132 . ПМИД 17016423 . S2CID 8872470 .
- ^ Андерсон AC (сентябрь 2003 г.). «Процесс структурного проектирования лекарств» . Химия и биология . 10 (9): 787–797. doi : 10.1016/j.chembiol.2003.09.002 . ПМИД 14522049 .
- ^ Реканатини М., Боттегони Дж., Кавалли А. (декабрь 2004 г.). «Антицелевой скрининг in silico». Открытие наркотиков сегодня. Технологии . 1 (3): 209–215. дои : 10.1016/j.ddtec.2004.10.004 . ПМИД 24981487 .
- ^ Ву-Понг С., Роджанасакул Ю. (2008). Проектирование и разработка биофармацевтических лекарств (2-е изд.). Тотова, Нью-Джерси Humana Press: Humana Press. ISBN 978-1-59745-532-9 .
- ^ Скомпарин А, Поляк Д, Кривицкий А, Сатчи-Файнаро Р (ноябрь 2015 г.). «Достижение успешной доставки олигонуклеотидов - от физико-химической характеристики до оценки in vivo». Достижения биотехнологии . 33 (6, часть 3): 1294–1309. doi : 10.1016/j.biotechadv.2015.04.008 . ПМИД 25916823 .
- ^ Юсеф М., Хитти С., Пуппин Чавес Фулбер Дж., Камен А.А. (октябрь 2023 г.). «Возможность применения мРНК-терапии: текущая ситуация и проблемы в производстве» . Биомолекулы . 13 (10): 1497. doi : 10.3390/biom13101497 . ПМЦ 10604719 . ПМИД 37892179 .
- ^ Сахин У, Карико К, Тюречи О (октябрь 2014 г.). «Терапия на основе мРНК – разработка нового класса лекарств» . Обзоры природы. Открытие наркотиков . 13 (10): 759–780. дои : 10.1038/nrd4278 . ПМИД 25233993 . S2CID 27454546 .
- ^ Суинни, округ Колумбия, Ли Дж.А. (2020). «Последние достижения в открытии фенотипических лекарств» . F1000Исследования . 9 : F1000 Факультет Ред. – 944. дои : 10.12688/f1000research.25813.1 . ПМЦ 7431967 . ПМИД 32850117 .
- ^ Моффат Дж.Г., Винсент Ф., Ли Дж.А., Эдер Дж., Прунотто М. (август 2017 г.). «Возможности и проблемы в открытии фенотипических лекарств: взгляд на индустрию» . Обзоры природы. Открытие наркотиков . 16 (8): 531–543. дои : 10.1038/nrd.2017.111 . ПМИД 28685762 . S2CID 6180139 .
- ^ Ганеллин С.Р., Джефферис Р., Робертс С.М. (2013). «Процесс открытия низкомолекулярных лекарств — от выбора цели до выбора кандидата» . Введение в исследования и разработки биологических и низкомолекулярных лекарств: теория и тематические исследования . Эльзевир. ISBN 9780123971760 .
- ^ Jump up to: а б с Юань Ю, Пей Дж, Лай Л (декабрь 2013 г.). «Обнаружение сайта связывания и прогнозирование лекарственной способности белковых мишеней для разработки лекарств на основе структуры». Текущий фармацевтический дизайн . 19 (12): 2326–2333. дои : 10.2174/1381612811319120019 . ПМИД 23082974 .
- ^ Риштон GM (январь 2003 г.). «Несвинцовость и сходство со свинцом в биохимическом скрининге». Открытие наркотиков сегодня . 8 (2): 86–96. дои : 10.1016/s1359644602025722 . ПМИД 12565011 .
- ^ Хопкинс А.Л. (2011). «Глава 25: Фармакологическое пространство» . В Вермуте CG (ред.). Практика медицинской химии (3-е изд.). Академическая пресса. стр. 521–527. ISBN 978-0-12-374194-3 .
- ^ Кирхмайр Дж (2014). Прогноз метаболизма лекарств . Методы и принципы Уайли в медицинской химии. Том. 63. Вайли-ВЧ. ISBN 978-3-527-67301-8 .
- ^ Николау, Калифорния, Браун Н. (сентябрь 2013 г.). «Методы многокритериальной оптимизации при разработке лекарств». Открытие наркотиков сегодня. Технологии . 10 (3): е427–е435. дои : 10.1016/j.ddtec.2013.02.001 . ПМИД 24050140 .
- ^ Бан ТА (2006). «Роль случайности в открытии лекарств» . Диалоги в клинической неврологии . 8 (3): 335–344. дои : 10.31887/DCNS.2006.8.3/tban . ПМК 3181823 . ПМИД 17117615 .
- ^ Этирадж С.К., Левинталь Д. (сентябрь 2004 г.). «Ограниченная рациональность и поиск организационной архитектуры: эволюционный взгляд на дизайн организаций и их способность к развитию» . Ежеквартальный журнал административной науки . 49 (3). Sage Publications, Inc. от имени Высшей школы менеджмента Джонсона Корнельского университета: 404–437. дои : 10.2307/4131441 . JSTOR 4131441 . S2CID 142910916 . ССНР 604123 .
- ^ Льюис Р.А. (2011). «Глава 4: Разработка программ молекулярного моделирования: использование и ограничения физических моделей». В Gramatica P, Ливингстон DJ, Дэвис AM (ред.). Стратегии разработки лекарств: количественные подходы . RSC Открытие лекарств. Королевское химическое общество. стр. 88–107. дои : 10.1039/9781849733410-00088 . ISBN 978-1849731669 .
- ^ Раджамани Р., Good AC (май 2007 г.). «Рейтинг представляет собой структурное обнаружение и оптимизацию потенциальных клиентов: текущие тенденции в развитии функции оценки». Текущее мнение об открытии и разработке лекарств . 10 (3): 308–315. ПМИД 17554857 .
- ^ де Азеведо В.Ф., Диас Р. (декабрь 2008 г.). «Вычислительные методы расчета аффинности связывания лиганда». Текущие цели по борьбе с наркотиками . 9 (12): 1031–1039. дои : 10.2174/138945008786949405 . ПМИД 19128212 .
- ^ Сингх Дж., Чуаки С.Э., Бориак-Сьодин П.А., Ли В.К., Понц Т., Корбли М.Дж. и др. (декабрь 2003 г.). «Успешный виртуальный скрининг на основе формы: открытие мощного ингибитора киназы рецептора TGFbeta типа I (TbetaRI)». Письма по биоорганической и медицинской химии . 13 (24): 4355–4359. дои : 10.1016/j.bmcl.2003.09.028 . ПМИД 14643325 .
- ^ Беккер О.М., Дханоа Д.С., Маранц Ю., Чен Д., Шахам С., Черуку С. и др. (июнь 2006 г.). «Интегрированное in silico 3D-модель открытие нового, мощного и селективного агониста амидосульфонамида 5-HT1A (PRX-00023) для лечения тревоги и депрессии». Журнал медицинской химии . 49 (11): 3116–3135. дои : 10.1021/jm0508641 . ПМИД 16722631 .
- ^ Лян С., Меруэ СО, Ван Г, Цю С, Чжоу Ю (май 2009 г.). «Консенсусная оценка для обогащения почти нативных структур из ловушек для стыковки белков с белками» . Белки . 75 (2): 397–403. дои : 10.1002/прот.22252 . ПМЦ 2656599 . ПМИД 18831053 .
- ^ Ода А., Цучида К., Такакура Т., Ямаоцу Н., Хироно С. (2006). «Сравнение стратегий консенсусной оценки для оценки вычислительных моделей белково-лигандных комплексов». Журнал химической информации и моделирования . 46 (1): 380–391. дои : 10.1021/ci050283k . ПМИД 16426072 .
- ^ Дэн З., Чуаки С., Сингх Дж. (январь 2004 г.). «Отпечаток структурного взаимодействия (SIFt): новый метод анализа трехмерных взаимодействий связывания белок-лиганд». Журнал медицинской химии . 47 (2): 337–344. дои : 10.1021/jm030331x . ПМИД 14711306 .
- ^ Амари С., Айзава М., Чжан Дж., Фукузава К., Мотидзуки Ю., Ивасава Ю. и др. (2006). «VISCANA: визуализированный кластерный анализ взаимодействия белок-лиганд на основе метода молекулярных орбиталей фрагментов ab initio для скрининга виртуальных лигандов». Журнал химической информации и моделирования . 46 (1): 221–230. дои : 10.1021/ci050262q . ПМИД 16426058 .
- ^ Гюнер О.Ф. (2000). Восприятие, разработка и использование фармакофоров при разработке лекарств . Ла-Хойя, Калифорния: Международная университетская линия. ISBN 978-0-9636817-6-8 .
- ^ Тропша А (2010). «QSAR в открытии лекарств» . В Рейнольдсе Ч., Мерце К.М., Ринге Д. (ред.). Разработка лекарств: подходы, основанные на структуре и лигандах (1-е изд.). Кембридж, Великобритания: Издательство Кембриджского университета. стр. 151–164. ISBN 978-0521887236 .
- ^ Лич А.Р., Харрен Дж. (2007). Открытие лекарств на основе структуры . Берлин: Шпрингер. ISBN 978-1-4020-4406-9 .
- ^ Маузер Х., Губа В. (май 2008 г.). «Последние разработки в области проектирования de novo и установки строительных лесов». Текущее мнение об открытии и разработке лекарств . 11 (3): 365–374. ПМИД 18428090 .
- ^ Jump up to: а б Клебе Г (2000). «Последние разработки в области разработки лекарств на основе структуры». Журнал молекулярной медицины . 78 (5): 269–281. дои : 10.1007/s001090000084 . ПМИД 10954199 . S2CID 21314020 .
- ^ Ван Р., Гао Ю, Лай Л. (2000). «LigBuilder: многоцелевая программа для разработки лекарств на основе структуры». Журнал молекулярного моделирования . 6 (7–8): 498–516. дои : 10.1007/s0089400060498 . S2CID 59482623 .
- ^ Шнайдер Г., Фехнер Ю. (август 2005 г.). «Компьютерный дизайн молекул, подобных лекарствам, de novo». Обзоры природы. Открытие наркотиков . 4 (8): 649–663. дои : 10.1038/nrd1799 . ПМИД 16056391 . S2CID 2549851 .
- ^ Йоргенсен В.Л. (март 2004 г.). «Множество ролей вычислений в открытии лекарств». Наука . 303 (5665): 1813–1818. Бибкод : 2004Sci...303.1813J . дои : 10.1126/science.1096361 . ПМИД 15031495 . S2CID 1307935 .
- ^ Jump up to: а б Лейс С., Шнайдер С., Захариас М. (2010). «In silico предсказание сайтов связывания белков». Современная медицинская химия . 17 (15): 1550–1562. дои : 10.2174/092986710790979944 . ПМИД 20166931 .
- ^ Уоррен Г.Л., Уоррен С.Д. (2011). «Глава 16: Оценка взаимодействий лекарственного средства и рецептора». В Gramatica P, Ливингстон DJ, Дэвис AM (ред.). Стратегии разработки лекарств: количественные подходы . RSC Открытие лекарств. Королевское химическое общество. стр. 440–457. дои : 10.1039/9781849733410-00440 . ISBN 978-1849731669 .
- ^ Бём Х.Дж. (июнь 1994 г.). «Разработка простой эмпирической оценочной функции для оценки константы связывания комплекса белок-лиганд известной трехмерной структуры». Журнал компьютерного молекулярного дизайна . 8 (3): 243–256. Бибкод : 1994JCAMD...8..243B . дои : 10.1007/BF00126743 . ПМИД 7964925 . S2CID 2491616 .
- ^ Лю Дж, Ван Р. (март 2015 г.). «Классификация текущих скоринговых функций». Журнал химической информации и моделирования . 55 (3): 475–482. дои : 10.1021/ci500731a . ПМИД 25647463 .
- ^ Мурко М.А. (декабрь 1995 г.). «Вычислительные методы прогнозирования свободной энергии связывания в комплексах лиганд-рецептор». Журнал медицинской химии . 38 (26): 4953–4967. дои : 10.1021/jm00026a001 . ПМИД 8544170 .
- ^ Граматика П (2011). «Глава 17: Моделирование химических веществ в окружающей среде» . В Gramatica P, Ливингстон DJ, Дэвис AM (ред.). Стратегии разработки лекарств: количественные подходы . RSC Открытие лекарств. Королевское химическое общество. п. 466. дои : 10.1039/9781849733410-00458 . ISBN 978-1849731669 .
- ^ Грир Дж., Эриксон Дж.В., Болдуин Дж.Дж., Варни, доктор медицинских наук (апрель 1994 г.). «Применение трехмерных структур белковых молекул-мишеней в структурном дизайне лекарств». Журнал медицинской химии . 37 (8): 1035–1054. дои : 10.1021/jm00034a001 . ПМИД 8164249 .
- ^ Тиммерман Х., Губернатор К., Бём Х.Дж., Маннхольд Р., Кубиный Х. (1998). Структурно-ориентированный дизайн лигандов (методы и принципы медицинской химии) . Вайнхайм: Wiley-VCH. ISBN 978-3-527-29343-8 .
- ^ Капдевиль Р., Бухдангер Э., Циммерман Дж., Matter A (июль 2002 г.). «Гливек (STI571, иматиниб), рационально разработанный таргетный противораковый препарат». Обзоры природы. Открытие наркотиков . 1 (7): 493–502. дои : 10.1038/nrd839 . ПМИД 12120256 . S2CID 2728341 .
- ^ «Роль AutoDock в разработке первого клинически одобренного ингибитора ВИЧ-интегразы» . Пресс-релиз . Исследовательский институт Скриппса. 17 декабря 2007 г.
- ^ Прайор М., Чирута С., Куррейс А., Голдберг Дж., Рэмси Дж., Даргуш Р. и др. (июль 2014 г.). «Назад в будущее с фенотипическим скринингом» . ACS Химическая нейронаука . 5 (7): 503–513. дои : 10.1021/cn500051h . ПМК 4102969 . ПМИД 24902068 .
- ^ Коц Дж. (апрель 2012 г.). «Фенотипический скрининг, дубль два» . Наука-Бизнес ОБМЕН . 5 (15): 380. doi : 10.1038/scibx.2012.380 . ISSN 1945-3477 . S2CID 72519717 .
- ^ Герцберг Р.П., Папа А.Дж. (август 2000 г.). «Высокопроизводительный скрининг: новая технология 21 века». Современное мнение в области химической биологии . 4 (4): 445–451. дои : 10.1016/S1367-5931(00)00110-1 . ПМИД 10959774 .
- ^ Уолтерс В.П., Шталь М.Т., Мурко М.А. (апрель 1998 г.). «Виртуальный скрининг - обзор». Открытие наркотиков сегодня . 3 (4): 160–178. дои : 10.1016/S1359-6446(97)01163-X .
- ^ Кляйн Д.Ф. (март 2008 г.). «Утрата случайности в психофармакологии». ДЖАМА . 299 (9): 1063–1065. дои : 10.1001/jama.299.9.1063 . ПМИД 18319418 .
Внешние ссылки
[ редактировать ]- Лекарство + дизайн в Национальной медицинской библиотеке США по медицинским предметным рубрикам (MeSH)
- [Организация по разработке лекарств]( https://www.drugdesign.org/chapters/drug-design/ )