Филадельфийская хромосома
| Филадельфийская хромосома | |
|---|---|
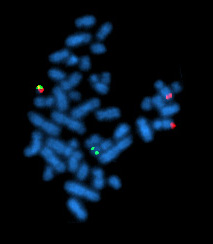 | |
| Метафазная клетка, положительная на перегруппировку bcr/abl с использованием FISH | |
| Специальность | онкология |
или Филадельфийская хромосома Филадельфийская транслокация ( Ph ) представляет собой специфическую генетическую аномалию в хромосоме 22 раковых клеток лейкемии (особенно клеток хронического миелолейкоза (ХМЛ)). Эта хромосома дефектна и необычно коротка из-за реципрокной транслокации t(9;22)(q34;q11) генетического материала между хромосомой 9 и хромосомой 22 и содержит слитый ген под названием BCR-ABL1 . Этот ген представляет собой ген ABL1 хромосомы 9, расположенный рядом с геном BCR области кластера точки разрыва хромосомы 22, кодирующим гибридный белок: белок тирозинкиназы сигнальный , который «всегда включен», заставляя клетку бесконтрольно делиться , нарушая стабильность генома и нарушая различные сигнальные пути, управляющие клеточным циклом. [1]
Наличие этой транслокации необходимо для диагностики ХМЛ; другими словами, все случаи ХМЛ положительны для BCR-ABL1 . [2] (Некоторые случаи смешиваются либо с загадочной транслокацией, которая невидима на препаратах G- хромосомы, либо с вариантной транслокацией, затрагивающей другую хромосому или хромосомы, а также длинное плечо хромосом 9 и 22. Другие подобные, но действительно Ph-отрицательные состояния: считаются ХМЛ-подобными миелопролиферативными новообразованиями. [3] ) Однако наличие Филадельфийской (Ph) хромосомы недостаточно специфично для диагностики ХМЛ, поскольку ее обнаруживают и при остром лимфобластном лейкозе. [4] (также известный как ОЛЛ, 25–30% случаев у взрослых и 2–10% случаев у детей ), а иногда и при остром миелогенном лейкозе (ОМЛ), а также остром лейкозе смешанного фенотипа (MPAL).
биология Молекулярная

Хромосомный дефект филадельфийской хромосомы представляет собой реципрокную транслокацию , при которой части двух хромосом, 9 и 22, меняются местами. В результате слитый ген создается ABL1 путем сопоставления гена на хромосоме 9 (область q34) с частью гена BCR (область кластера точек разрыва) на хромосоме 22 (область q11). Это реципрокная транслокация, в результате которой образуется удлиненная хромосома 9 (называемая производной хромосомой, или дер 9 ) и укороченная хромосома 22 ( филадельфийская хромосома, 22q-). [5] [6] В соответствии с Международной системой цитогенетической номенклатуры человека (ISCN) эта хромосомная транслокация обозначается как t(9;22)(q34;q11). Символ ABL1 происходит от Abelson — названия вируса лейкемии , несущего аналогичный белок. Символ BCR происходит от области кластера точек разрыва, гена, который кодирует белок, который действует как фактор обмена гуаниновых нуклеотидов для белков Rho GTPase. [7]
Транслокация приводит к слиянию онкогенных генов BCR-ABL1 , которое можно обнаружить на более короткой производной хромосоме 22. Этот ген кодирует слитый белок BCR-ABL1. В зависимости от точного места слияния молекулярная масса этого белка может составлять от 185 до 210 кДа . Следовательно, гибридный слитый белок BCR-ABL1 обозначается как p210 или p185.
Три клинически важных варианта, кодируемых слитым геном, представляют собой изоформы p190, p210 и p230. [8] p190 обычно связан с острым B-клеточным лимфобластным лейкозом (ОЛЛ), тогда как р210 обычно связан с хроническим миелолейкозом , но также может быть связан с ОЛЛ и ОМЛ. [9] p230 обычно связан с хроническим миелогенным лейкозом, ассоциированным с нейтрофилией и тромбоцитозом (ХМЛ-Н). [9] Кроме того, изоформа p190 также может экспрессироваться как сплайсинговый вариант p210. [10]
Ген ABL1 экспрессирует мембраносвязанный белок, тирозинкиназу , а транскрипт BCR-ABL1 также транслируется в тирозинкиназу, содержащую домены как из генов BCR , так и из ABL1 . Активность тирозинкиназ обычно регулируется автоингибирующим способом, но слитый ген BCR-ABL1 кодирует белок, который «всегда включен» или конститутивно активирован, что приводит к нарушению связывания ДНК и нерегулируемому делению клеток (т.е. к раку). Это происходит из-за замены миристоилированной кэп-области, которая, когда она присутствует, вызывает конформационные изменения, делающие киназный домен неактивным, на укороченную часть белка BCR. [11] Хотя область BCR также экспрессирует серин/треониновые киназы, функция тирозинкиназы очень важна для лекарственной терапии. Поскольку N-концевые домены Y177 и CC BCR кодируют конститутивную активацию киназы ABL1, эти области используются в терапии для подавления активности киназы BCR-ABL1. Ингибиторы тирозинкиназы, специфичные для таких доменов, как CC, Y177 и Rho (такие как иматиниб и сунитиниб ), являются важными лекарственными средствами против различных видов рака, включая ХМЛ, почечно-клеточный рак (ПКР) и стромальные опухоли желудочно-кишечного тракта (ГИСО).
Слитый белок BCR-ABL1 взаимодействует с субъединицей бета(с) рецептора интерлейкина-3 и регулируется петлей активации внутри его домена SH1, которая включается при связывании с АТФ и запускает нижестоящие пути. Тирозинкиназная активность ABL1 BCR-ABL1 повышена по сравнению с ABL1 дикого типа. [12] Поскольку ABL активирует ряд клеточный цикл , контролирующих белков и ферментов , результатом слияния BCR-ABL1 является ускорение деления клеток. Более того, он ингибирует репарацию ДНК , вызывая геномную нестабильность и потенциально вызывая опасный бластный кризис при ХМЛ.
при Пролиферативная роль лейкемии
Слитый ген и белок BCR-ABL1, кодируемые филадельфийской хромосомой, влияют на множество сигнальных путей, которые напрямую влияют на апоптотический потенциал, скорость деления клеток и различные стадии клеточного цикла, обеспечивая неконтролируемую пролиферацию, характерную для ХМЛ и ОЛЛ.
Путь JAK/STAT [ править ]
Особенно важным для выживания и пролиферации клеток миелолейкоза в микроокружении костного мозга является передача сигналов цитокинов и факторов роста. Путь JAK/STAT смягчает многие из этих эффекторов путем активации STAT, которые являются факторами транскрипции, способными модулировать рецепторы цитокинов и факторы роста. JAK2 фосфорилирует слитый белок BCR-ABL по Y177 и стабилизирует слитый белок, усиливая передачу сигналов онкогенных клеток. Было показано, что мутации JAK2 играют центральную роль в миелопролиферативных новообразованиях, а киназы JAK играют центральную роль в развитии гематологических злокачественных новообразований (журнал крови JAK). Терапия ALL и CML нацелена на JAK2, а также BCR-ABL с использованием нилотиниба и руксолитиниба на мышиных моделях для подавления последующей передачи сигналов цитокинов путем подавления активации транскрипции STAT3 и STAT5 (appelmann et al.). Взаимодействие между JAK2 и BCR-ABL в рамках этих гемопоэтических злокачественных опухолей предполагает важную роль JAK-STAT-опосредованной передачи сигналов цитокинов в стимулировании роста лейкозных клеток, проявляющих Ph-хромосому и тирозинкиназную активность BCR-ABL. Хотя центральное значение пути JAK2 для прямой пролиферации при ХМЛ обсуждается, его роль как нижестоящего эффектора тирозинкиназы BCR-ABL сохраняется. Воздействия на клеточный цикл через JAK-STAT в основном являются периферическими, но, непосредственно влияя на поддержание кроветворной ниши и окружающего ее микроокружения, активация BCR-ABL передачи сигналов JAK-STAT играет важную роль в поддержании роста и деления лейкозных клеток. [13] [14]
Путь Ras/ ERK / MAPK
Путь Ras/MAPK/ERK передает сигналы к факторам ядерной транскрипции и играет роль в управлении контролем и дифференцировкой клеточного цикла. В клетках, содержащих Ph-хромосому, тирозинкиназа BCR-ABL активирует путь RAS/RAF/MEK/ERK, что приводит к нерегулируемой пролиферации клеток посредством транскрипции генов в ядре. Тирозинкиназа BCR-ABL активирует Ras посредством фосфорилирования белка GAB2, которое зависит от фосфорилирования Y177, локализованного в BCR. В частности, показано, что Ras является важной последующей мишенью BCR-ABL1 при ХМЛ, поскольку мутанты Ras в мышиных моделях нарушают развитие ХМЛ, связанного с геном BCR-ABL1 (эффект ингибирования Ras в гемопоэзе и лейкемогенезе BCR/ABL). Путь Ras/RAF/MEK/ERK также участвует в сверхэкспрессии остеопонтина (OPN), который важен для поддержания ниши гемопоэтических стволовых клеток, что косвенно влияет на неконтролируемую пролиферацию, характерную для лейкозных клеток. [15] Слитые клетки BCR-ABL также демонстрируют конститутивно высокие уровни активированного Ras, связанного с GTP, активируя Ras-зависимый сигнальный путь, который, как было показано, ингибирует апоптоз ниже BCR-ABL (Cortez et al.). Взаимодействие с рецептором IL-3 также индуцирует путь Ras/RAF/MEK/ERK для фосфорилирования факторов транскрипции, которые играют роль в управлении переходом G1/S клеточного цикла. [16] [17] [18]
ДНК апоптоз Связывание и
Ген c-Abl в клетках дикого типа участвует в связывании ДНК, что влияет на такие процессы, как транскрипция ДНК, репарация, апоптоз и другие процессы, лежащие в основе клеточного цикла. Хотя природа этого взаимодействия обсуждается, существуют данные, позволяющие предположить, что c-Abl фосфорилирует HIPK2 , серин/треониновую киназу, в ответ на повреждение ДНК и способствует апоптозу в нормальных клетках. Напротив, было показано, что слияние BCR-ABL ингибирует апоптоз, но его влияние, в частности, на связывание ДНК, неясно. [19] Было показано, что при апоптотическом ингибировании клетки BCR-ABL устойчивы к апоптозу, индуцированному лекарственными средствами, но также имеют проапоптотический профиль экспрессии за счет повышенных уровней экспрессии p53, p21 и Bax. Однако функция этих проапоптотических белков нарушена, и апоптоз в этих клетках не осуществляется. BCR-ABL также участвует в предотвращении процессинга каспазы 9 и каспазы 3, что усиливает ингибирующий эффект. [20] [21] Другим фактором, предотвращающим прогрессирование клеточного цикла и апоптоз, является делеция гена IKAROS , которая присутствует в более чем 80% случаев ОЛЛ с Ph-хромосомой. Ген IKAROS имеет решающее значение для остановки клеточного цикла, опосредованной пре-В-клеточным рецептором, во ВСЕХ клетках, положительных по Ph, который при нарушении обеспечивает механизм неконтролируемого прогрессирования клеточного цикла и пролиферации дефектных клеток, что стимулируется передачей сигналов тирозинкиназы BCR-ABL. [22]
Номенклатура [ править ]
Филадельфийская хромосома обозначается Ph (или Ph') хромосомой и обозначает укороченную хромосому 22, которая кодирует слитый ген BCR-ABL/протеинкиназу. Это возникает в результате транслокации, которая называется t(9;22)(q34.1;q11.2) между хромосомой 9 и хромосомой 22, с разрывами, происходящими в области (3), полосе (4), поддиапазоне ( 1) длинного плеча (q) хромосомы 9 и области (1), полосы (1), поддиапазона (2) длинного плеча (q) хромосомы 22. Следовательно, точки разрыва хромосомы записываются как (9q34. 1) и (22q11.2) соответственно, с использованием стандартов ISCN.
Терапия [ править ]
Ингибиторы тирозинкиназы [ править ]

В конце 1990-х годов STI-571 ( иматиниб , Gleevec/Glivec) был идентифицирован фармацевтической компанией Novartis (тогда известной как Ciba Geigy) в ходе высокопроизводительного скрининга ингибиторов тирозинкиназы . Последующие клинические испытания под руководством доктора Брайана Дж. Друкера из Орегонского университета здоровья и науки в сотрудничестве с доктором Чарльзом Сойерсом и доктором Моше Талпазом продемонстрировали, что STI-571 ингибирует пролиферацию гемопоэтических клеток, экспрессирующих BCR-ABL. Хотя он и не уничтожил клетки ХМЛ, он значительно ограничил рост опухолевого клона и снизил риск опасного « взрывного кризиса ». [ нужна ссылка ] В 2000 году доктор Джон Куриян определил механизм, с помощью которого STI-571 ингибирует киназный домен Abl. [23] В 2001 году компания Novartis продавала его под названием мезилат иматиниба (Гливек в США, Гливек в Европе).
Разрабатываются другие фармакологические ингибиторы, которые являются более мощными и/или активными против появляющихся устойчивых к Гливеку/Гливеку клонов BCR-abl у пролеченных пациентов. Большинство этих устойчивых клонов представляют собой точечные мутации киназы BCR-abl. Новые ингибиторы включают дазатиниб и нилотиниб , которые значительно более эффективны, чем иматиниб, и могут преодолевать резистентность. Комбинированная терапия нилотинибом и руксолитнибом также показала успех в подавлении резистентности, одновременно воздействуя на стадии JAK-STAT и BCR-ABL. Низкомолекулярные ингибиторы, такие как триоксид мышьяка и аналоги гелданамицина , также были идентифицированы как подавляющие трансляцию киназы BCR-ABL и способствующие ее деградации протеазой. [24] [25]
Было показано, что акситиниб , препарат, используемый для лечения почечно-клеточного рака, эффективен в ингибировании активности киназы Abl у пациентов с BCR-ABL1 (T315I). [26] Мутация T315I в слитом гене обеспечивает устойчивость к другим ингибиторам тирозинкиназы, таким как иматиниб, однако акситиниб успешно использовался для лечения пациентов с ОЛЛ, несущих эту мутацию, а также клеток ХМЛ в культуре.
Лечение Ph+ ОЛЛ у детей комбинацией стандартной химиотерапии и ингибиторов РТК может привести к ремиссии. [ нужна ссылка ] но лечебный потенциал неизвестен.
Ациминиб (Сцембликс) был одобрен для медицинского применения в США в октябре 2021 года. [27]
крови или Трансплантация мозга костного
Потенциально излечивающим, но рискованным вариантом лечения Ph+ ALL или Ph+ CML у детей является трансплантация костного мозга или трансплантация пуповинной крови , но некоторые отдают предпочтение химиотерапии для достижения первой ремиссии (CR1). Для некоторых трансплантация костного мозга от подходящего донора-брата или сестры или подходящего неродственного донора может быть предпочтительной при достижении ремиссии.
Некоторые предпочитают трансплантацию пуповинной крови , когда совпадение костного мозга 10/10 недоступно, и трансплантация пуповинной крови может иметь некоторые преимущества, в том числе снижение частоты реакции «трансплантат против хозяина» (РТПХ), которая является распространенным и серьезным осложнением. трансплантата. Однако трансплантация пуповинной крови иногда требует более длительного периода времени для приживления, что может увеличить вероятность осложнений из-за инфекции. Независимо от типа трансплантата, возможны смертность и рецидивы, связанные с трансплантацией, и эти показатели могут меняться по мере совершенствования протоколов лечения. Для второй ремиссии (CR2), если она достигнута, возможны варианты как химиотерапии, так и трансплантации, и многие врачи предпочитают трансплантацию. [ нужна ссылка ]
Прогноз [ править ]
Согласно исследованиям эпохи ингибиторов тирозинкиназы, BCR-ABL-положительный острый лимфобластный лейкоз (ОЛЛ) имеет 5-летнюю выживаемость в диапазоне от 50% до 75%. [28]
История [ править ]
Филадельфийская хромосома была впервые обнаружена и описана в 1959 году Дэвидом Хангерфордом в Институте исследования рака при больнице Ланкенау , который в 1974 году объединился с Американской онкологической больницей и создал онкологический центр Фокса Чейза . [29] вместе с Питером Ноуэллом из Медицинской школы Пенсильванского университета . Генетическая аномалия, обнаруженная Хангерфордом и Ноуэллом, была названа в честь города, в котором располагались обе организации. [1] [30] [29] [31] И таким образом, это типичный пример медицинского топонима .
Хангерфорд писал докторскую диссертацию по хромосомам в генетической лаборатории тогдашнего Института исследований рака при Научно-исследовательском институте больницы Ланкенау. [29] и обнаружил дефект в хромосомах клеток крови пациентов с лейкемией. Это основополагающее наблюдение стало первым генетическим дефектом, связанным с конкретным раком человека. Ноуэлл был патологом из Пенсильванского университета, который также изучал клетки лейкемии под микроскопом, когда заметил клетки с этим генетическим дефектом в процессе деления. К его удивлению, их хромосомы — обычно нечеткие клубки — были видны как отдельные структуры. В поисках эксперта по хромосомам Ноуэлл нашел Хангерфорда в Ланкенау. Проводя микроскопические исследования, Хангерфорд продолжил свои наблюдения, обнаружив, что некоторые лейкозные клетки имеют аномально короткую 22-ю хромосому. Впоследствии обнаруженная им мутация стала известна как Филадельфийская хромосома.
В 1973 году Джанет Роули из Чикагского университета определила механизм возникновения филадельфийской хромосомы в виде транслокации. [1] [32] [33]
См. также [ править ]
Ссылки [ править ]
- ↑ Перейти обратно: Перейти обратно: а б с Вапнер Дж. (2014). Филадельфийская хромосома: генетическая загадка, смертельный рак и невероятное изобретение лекарства, спасающего жизнь . Нью-Йорк, штат Нью-Йорк: Эксперимент. ISBN 978-1-61519-197-0 .
- ^ «Хронический миелолейкоз» (PDF) . Рекомендации NCCN по клинической практике . Национальная комплексная сеть по борьбе с раком. 15 ноября 2021 г. Проверено 15 июля 2020 г.
- ^ «Миелопролиферативные новообразования» (PDF) . Рекомендации NCCN по клинической практике . Национальная комплексная сеть по борьбе с раком. 15 ноября 2021 г. Проверено 20 февраля 2018 г.
- ^ Талпаз М., Шах Н.П., Кантарджян Х., Донато Н., Николл Дж., Пакетт Р. и др. (июнь 2006 г.). «Дазатиниб при резистентных к иматинибу лейкозах с филадельфийской хромосомой» . Медицинский журнал Новой Англии . 354 (24): 2531–2541. doi : 10.1056/NEJMoa055229 . ПМИД 16775234 .
- ^ Курцрок Р., Кантарджян Х.М., Друкер Б.Дж., Талпаз М. (май 2003 г.). «Лейкозы с положительной филадельфийской хромосомой: от основных механизмов к молекулярной терапии». Анналы внутренней медицины . 138 (10): 819–830. дои : 10.7326/0003-4819-138-10-200305200-00010 . ПМИД 12755554 . S2CID 25865321 .
- ^ Мело СП (май 1996 г.). «Молекулярная биология хронического миелолейкоза». Лейкемия . 10 (5): 751–756. ПМИД 8656667 .
- ^ «Запись гена для BCR» . NCBI Ген . Проверено 21 января 2020 г.
- ^ Адвани А.С., Пендергаст А.М. (август 2002 г.). «Варианты Bcr-Abl: биологические и клинические аспекты» . Исследования лейкемии . 26 (8): 713–720. дои : 10.1016/s0145-2126(01)00197-7 . ПМИД 12191565 .
- ↑ Перейти обратно: Перейти обратно: а б Пакакасама С., Каджаначумпол С., Канжанапонгкул С., Сирачайнан Н., Микаевкунчорн А., Нингсанонд В., Хонгэн С. (август 2008 г.). «Простая мультиплексная ОТ-ПЦР для выявления общих транскриптов слияния при остром лейкозе у детей» . Международный журнал лабораторной гематологии . 30 (4): 286–291. дои : 10.1111/j.1751-553X.2007.00954.x . ПМИД 18665825 .
- ^ Личти Б.Д., Китинг А., Каллум Дж., Йи К., Кроксфорд Р., Корпус G и др. (декабрь 1998 г.). «Экспрессия p210 и p190 BCR-ABL вследствие альтернативного сплайсинга при хроническом миелолейкозе» . Британский журнал гематологии . 103 (3): 711–715. дои : 10.1046/j.1365-2141.1998.01033.x . ПМИД 9858221 .
- ^ Нагар Б., Ханчел О., Янг М.А., Шеффзек К., Вич Д., Борнманн В. и др. (март 2003 г.). «Структурная основа аутоингибирования тирозинкиназы c-Abl» . Клетка . 112 (6): 859–871. дои : 10.1016/s0092-8674(03)00194-6 . ПМИД 12654251 .
- ^ Саттлер М., Гриффин Джей.Д. (апрель 2001 г.). «Механизмы трансформации онкогеном BCR/ABL». Международный журнал гематологии . 73 (3): 278–291. дои : 10.1007/BF02981952 . ПМИД 11345193 . S2CID 20999134 .
- ^ Варш В., Вальц С., Сексл В. (сентябрь 2013 г.). «JAK на все руки: JAK2-STAT5 как новые терапевтические мишени при хроническом миелолейкозе BCR-ABL1+» . Кровь . 122 (13): 2167–2175. дои : 10.1182/blood-2014-04-567289 . ПМИД 23926299 .
- ^ Ханчель О (февраль 2015 г.). «Нацеливание на BCR-ABL и JAK2 в Ph+ ALL» . Кровь . 125 (9): 1362–1363. дои : 10.1182/blood-2014-12-617548 . ПМИД 25721043 .
- ^ Хейлок Д.Н., Нильссон С.К. (сентябрь 2006 г.). «Остеопонтин: мост между костью и кровью» . Британский журнал гематологии . 134 (5): 467–474. дои : 10.1111/j.1365-2141.2006.06218.x . ПМИД 16848793 .
- ^ Bandyopadhyay G, Biswas T, Roy KC, Mandal S, Mandal C, Pal BC и др. (октябрь 2004 г.). «Хлорогеновая кислота ингибирует тирозинкиназу Bcr-Abl и запускает митоген-активируемую протеинкиназой p38 апоптоз в клетках хронического миелогенного лейкоза» . Кровь . 104 (8): 2514–2522. дои : 10.1182/кровь-2003-11-4065 . ПМИД 15226183 .
- ^ Скорски Т., Канакарай П., Ку Д.Х., Неборовска-Скорска М., Канаани Э., Зон Г. и др. (июнь 1994 г.). «Негативная регуляция активности p120GAP GTPase, стимулирующей активность p210bcr / abl: значение для RAS-зависимого роста положительных клеток филадельфийской хромосомы» . Журнал экспериментальной медицины . 179 (6): 1855–1865. дои : 10.1084/jem.179.6.1855 . ПМК 2191514 . ПМИД 8195713 .
- ^ Стилман Л.С., Понерт С.С., Шелтон Дж.Г., Франклин Р.А., Бертран Ф.Е., МакКубри Дж.А. (февраль 2004 г.). «JAK/STAT, Raf/MEK/ERK, PI3K/Akt и BCR-ABL в прогрессировании клеточного цикла и лейкемогенезе» . Лейкемия . 18 (2): 189–218. дои : 10.1038/sj.leu.2403241 . ПМИД 14737178 .
- ^ Берк Б.А., Кэрролл М. (июнь 2010 г.). «BCR-ABL: многогранный промотор мутации ДНК при хроническом миелогенном лейкозе» . Лейкемия . 24 (6): 1105–1112. дои : 10.1038/leu.2010.67 . ПМЦ 4425294 . ПМИД 20445577 .
- ^ «Тирозинкиназа c-Abl реагирует на повреждение ДНК, активируя гомеодомен-взаимодействующую протеинкиназу 2» . Журнал биологической химии . 290 (27): 16489. 2015. doi : 10.1074/jbc.p114.628982 . ПМЦ 4505403 .
- ^ Кипреос ET, Ван JY (апрель 1992 г.). «Регулируемое клеточным циклом связывание тирозинкиназы c-Abl с ДНК». Наука . 256 (5055): 382–385. Бибкод : 1992Sci...256..382K . дои : 10.1126/science.256.5055.382 . ПМИД 1566087 . S2CID 29228735 .
- ^ Кази С., Укун FM (декабрь 2013 г.). «Частота и биологическое значение делеций гена IKZF1 / Ikaros у детей с отрицательным по Филадельфийской хромосоме и положительным по Филадельфийской хромосоме В-клеточным предшественником острого лимфобластного лейкоза» . Гематологическая . 98 (12): e151–e152. дои : 10.3324/haematol.2013.091140 . ПМЦ 3856976 . ПМИД 24323986 .
- ^ Шиндлер Т., Борнманн В., Пеллисена П., Миллер В.Т., Кларксон Б., Куриян Дж. (сентябрь 2000 г.). «Структурный механизм ингибирования STI-571 тирозинкиназы Абельсона». Наука . 289 (5486): 1938–1942. Бибкод : 2000Sci...289.1938S . дои : 10.1126/science.289.5486.1938 . ПМИД 10988075 . S2CID 957274 .
- ^ Нимманапалли Р., Бхалла К. (декабрь 2002 г.). «Новые таргетные методы лечения острых лейкозов, положительных по Bcr-Abl: помимо STI571» . Онкоген . 21 (56): 8584–8590. дои : 10.1038/sj.onc.1206086 . ПМИД 12476305 .
- ^ Дэн С., Найто М., Цуруо Т. (август 1998 г.). «Селективная индукция апоптоза в клетках хронического миелолейкоза с филадельфийской хромосомой ингибитором BCR - тирозинкиназы ABL, CGP 57148» . Смерть клеток и дифференцировка . 5 (8): 710–715. дои : 10.1038/sj.cdd.4400400 . ПМИД 10200527 .
- ^ Пемовска Т., Джонсон Э., Контро М., Репаски Г.А., Чен Дж., Уэллс П. и др. (март 2015 г.). «Акситиниб эффективно ингибирует BCR-ABL1 (T315I) с отчетливой конформацией связывания». Природа . 519 (7541): 102–105. Бибкод : 2015Natur.519..102P . дои : 10.1038/nature14119 . ПМИД 25686603 . S2CID 4389086 .
- ^ «FDA одобрило Novartis Schemblix (асциминиб) с новым механизмом действия для лечения хронического миелолейкоза» . Новартис (Пресс-релиз) . Проверено 29 октября 2021 г.
- ^ Леони В., Бионди А. (2015). «Ингибиторы тирозинкиназы при BCR-ABL-положительном остром лимфобластном лейкозе» . Гематологическая . 100 (3): 295–9. дои : 10.3324/haematol.2015.124016 . ПМЦ 4349266 . ПМИД 25740105 .
- ↑ Перейти обратно: Перейти обратно: а б с «История и свершения» . Институт медицинских исследований Ланкенау .
- ^ Онкологический центр Фокса Чейза (3 декабря 2015 г.). «50 лет открытия Филадельфийской хромосомы» . Архивировано из оригинала 02 апреля 2016 г. Проверено 28 июня 2011 г.
{{cite journal}}: Для цитирования журнала требуется|journal=( помощь ) - ^ «Национальная академия наук». Наука . 132 (3438): 1488–1501. Ноябрь 1960 г. Бибкод : 1960Sci...132.1488. . дои : 10.1126/science.132.3438.1488 . ПМИД 17739576 .
- ^ Роули JD (июнь 1973 г.). «Письмо: новая последовательная хромосомная аномалия при хроническом миелогенном лейкозе, выявленная с помощью хинакринной флуоресценции и окрашивания по Гимзе». Природа . 243 (5405): 290–293. Бибкод : 1973Natur.243..290R . дои : 10.1038/243290a0 . ПМИД 4126434 . S2CID 26726071 .
- ^ Дрейфус К. (07 февраля 2011 г.). «Матриарх современной генетики рака» . Нью-Йорк Таймс .
Внешние ссылки [ править ]
- Филадельфия + хромосома в Национальной медицинской библиотеке США по медицинским предметным рубрикам (MeSH)
- bcr-abl+Fusion+Proteins в Национальной медицинской библиотеке США по медицинским предметным рубрикам (MeSH)