Фолликулярная лимфома
| Фолликулярная лимфома | |
|---|---|
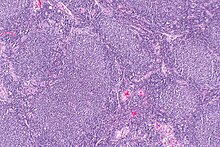 | |
| Микрофотография фолликулярной лимфомы, показывающая характерные аномальные лимфоидные фолликулы , которые и дали этому заболеванию название. Пятно H&E . | |
| Специальность | Гематология и онкология |
Фолликулярная лимфома ( ФЛ ) – это рак , который поражает определенные типы лейкоцитов, известных как лимфоциты . Рак возникает в результате неконтролируемого деления определенных типов B-клеток, известных как центроциты и центробласты . Эти клетки обычно занимают фолликулы (узловые завитки различных типов лимфоцитов) в зародышевых центрах лимфоидных тканей, таких как лимфатические узлы . Раковые клетки при ФЛ обычно образуют фолликулярные или фолликулоподобные структуры (см. рисунок рядом) в тканях, в которые они проникают. Эти структуры обычно являются доминирующей гистологической особенностью этого рака. [1]
Существует несколько синонимичных и устаревших терминов для ФЛ, таких как CB/CC-лимфома (центробластная и центроцитарная лимфома), узловая лимфома, [2] Болезнь Брилла-Симмерса и обозначение подтипа — фолликулярная крупноклеточная лимфома. [3] В США и Европе это заболевание является второй по распространенности формой неходжкинских лимфом , уступая только диффузной крупноклеточной В-клеточной лимфоме . [4] На долю ФЛ приходится 10–20% неходжкинских лимфом, при этом ежегодно в США и Европе диагностируется около 15 000 новых случаев. [5] Недавние исследования показывают, что ФЛ также распространена в Японии. [6]
ФЛ представляет собой широкую и чрезвычайно сложную клиническую единицу с широким спектром проявлений. [7] которые еще не полностью систематизированы. [8] Ему обычно предшествует доброкачественное предраковое заболевание , при котором аномальные центроциты и/или центробласты накапливаются в лимфоидной ткани. Затем они могут циркулировать в крови, вызывая бессимптомное состояние, называемое in situ лимфоидной неоплазией типа фолликулярной лимфомы (т.е. ИСФЛ). Небольшой процент этих случаев переходит в ФЛ. [9] Однако чаще всего ФЛ проявляется как увеличение лимфатических узлов на шее, подмышках и/или в паху. Реже он проявляется раком желудочно-кишечного тракта , раком у детей, поражающим лимфоидные ткани области головы и шеи (например, миндалины ), [10] или одно или несколько образований в нелимфоидных тканях, таких как семенники . [11]
ФЛ обычно имеет медленное течение заболевания, которое сохраняется практически без изменений в течение многих лет. [7] Однако ежегодно 2–3% [12] случаев ФЛ переходят в высокоагрессивную форму, часто называемую ФЛ стадии 3В, в агрессивную диффузную крупноклеточную В-клеточную лимфому или в другой тип агрессивного В-клеточного рака. Эти трансформированные фолликулярные лимфомы (т-ФЛ) практически неизлечимы. [5] Однако недавние достижения в лечении Т-ФЛ (например, добавление к стандартной химиотерапии таких препаратов, как ритуксимаб ) улучшили общее время выживаемости. Эти новые схемы могут также задерживать трансформацию ФЛ в т-ФЛ. [5] Дополнительные достижения в понимании ФЛ могут привести к дальнейшим улучшениям в лечении этого заболевания. [12] [13]
Патофизиология
[ редактировать ]Геномные изменения
[ редактировать ]Серийное прогрессирование in situ FL в FL и FL в t-FL, по-видимому, связано с накоплением возрастающего количества геномных изменений (т.е. хромосомных аномалий и генных мутаций ) в формирующих B-клетках-предшественниках этих нарушений. По крайней мере, некоторые из этих изменений, по-видимому, вызывают сверхэкспрессию или недостаточную экспрессию продуктов генов, которые регулируют восприимчивость этих клеток к развитию дальнейших геномных изменений , выживанию, пролиферации и/или распространению в другие ткани. В результате этого расстройства заселяют множественные клоны В-клеток, которые демонстрируют возрастающие геномные изменения и злокачественное поведение. Кажется, ни одно геномное изменение не несет ответственности за развитие каждого из спектров расстройств ФЛ. Скорее всего, в основе этой последовательной прогрессии лежат взаимодействия между множественными геномными изменениями. [5] [12]
in situ Фолликулярная лимфома
[ редактировать ]Фолликулярная лимфома in situ представляет собой скопление моноклональных В-клеток (т.е. клеток, происходящих от одной предковой клетки) в зародышевых центрах лимфоидной ткани. Эти клетки обычно несут патологическую геномную аномалию, т.е. транслокацию между позицией 32 на длинном (т.е. «q») плече хромосомы 14 и позицией 21 на q-плече хромосомы 18. Эта транслокация сопоставляет ген B-клеточной лимфомы 2 ( BCL2 ) на хромосоме 18 в положении q21.33 рядом с локусом тяжелой цепи иммуноглобулина ( IGH@ ) на хромосоме 14 в положении q32. В результате BCL2 сверхэкспрессирует свой продукт, регулятор апоптоза BCL2 (т.е. Bcl2). Bcl2 подавляет запрограммированную гибель клеток , тем самым продлевая выживаемость клеток. [14] Сверхэкспрессия Bcl2 в B-клетках ISFL считается критическим фактором их патологического накопления и последующего злокачественного прогрессирования. [9] Небольшое количество (например, 1 на 100 000) циркулирующих ядросодержащих клеток крови, несущих транслокацию t(14:18)q32:q21), обнаруживается у 50–67% здоровых людей. Распространенность этого явления увеличивается с возрастом и продолжительностью курения табака. Поскольку у большинства людей с этой транслокацией в клетках крови не развивается ИСФЛ, транслокация t(14:18)(q32:q21), хотя и продлевает выживаемость клеток, должна быть лишь одним шагом в развитии ИСФН. Предполагается, что эта транслокация происходит во время раннего развития незрелых B-клеток костного мозга (т.е. пре-B-клеток/про-B-клеток), после чего эти клетки свободно циркулируют и в редких случаях накапливаются и созревают до центроцитов и/или центробластов. в зародышевых центрах лимфоидных фолликулов с образованием ИСФЛ. Механизм, способствующий такой локализации и дальнейшему накоплению, неясен. [15]
У лиц с ИСЛЛ прогрессирование до ФЛ происходит со скоростью 2–3% в год в течение как минимум первых 10 лет после постановки диагноза. [12] Это прогрессирование, вероятно, связано с приобретением геномных аберраций помимо транслокации t(14:18)q32:q21) в B-клетках ISFL. Подозреваемые мутации включают мутации в следующих генах: 1) EZH2 (кодирует белок семейства полисотового репрессивного комплекса 2 , который участвует в поддержании транскрипционно- репрессивного состояния различных генов). [16] и встречается до 27% случаев ФЛ); [9] 2) CREBBP (кодирует CREB-связывающий белок, который способствует активации различных генов [17] ); 3) TNFSF14 (кодирует члена 14 суперсемейства факторов некроза опухоли, члена суперсемейства факторов некроза опухоли , который может функционировать как костимулирующий фактор для активации лимфоидных клеток [1] [18] ); и 4) KMT2D (кодирует гистон-лизин N-метилтрансферазу 2D, гистон-метилтрансферазу , которая регулирует экспрессию различных генов). [19] ). [20] ISFL также может приобретать многочисленные вариации числа копий (т.е. дупликации и делеции части хромосомы вместе с любым из содержащихся в ней генов), которые могут способствовать развитию FL. Во всех случаях количество приобретенных генетических аномалий в В-клетках ИСЛЛ значительно меньше, чем при ФЛ. [9]
Фолликулярная лимфома
[ редактировать ]Геномные изменения, обнаруженные при ФЛ, включают : 1) транслокацию t(14:18)(q32:q21.3) (85–90% случаев); 2) делеции 1p36 (т.е. делеции в плече q хромосомы 1 в положении 36, [60–70% случаев]), которые приводят к потере TNFAIP3 (кодирует фактор некроза опухоли, альфа-индуцированный белок 3, который ингибирует активацию NF -κB блокирует гибель клеток вследствие апоптоза и регулирует иммунные реакции лимфоцитов посредством убиквитинлигазы. активности [21] ); 3) мутации PRDM1 (кодирует белок цинкового пальца домена PR, который способствует созреванию и пролиферации В-клеток); [22] и 4) те же мутации, что и при ISFL, включая KMT2D (85–90% случаев), CREEBP (40–65% случаев), BCL2 (40–65% случаев) и EZH2 (20–30% случаев). а также другие мутации, такие как мутации в гене, модифицирующем гистоны HIST1H1E (20–30% случаев), гене RRAGC (~ 17% случаев), который регулирует рост, выживание, гибель и пролиферацию клеток, [23] и в ≤15% случаев несколько других генов, включая MEF2B , STAT6 , EP300 , ARID1A , SLC22A2 , CARD11 , FOXO1 , GNA12 , B2M (т.е. ген бета-2-микроглобулина ) и SGK1 . За исключением транслокации t(14:18)(q32:q21.3) и мутаций EZH2 , которые приводят к усилению экспрессии и функции их продуктов соответственно, генетические изменения обычно приводят к потере продукции или функции указанные продукты генов. Однако точная роль (если таковая имеется) этих геномных аномалий в содействии прогрессированию ИСЛЛ в ФЛ неясна. [24]
Трансформированная фолликулярная лимфома
[ редактировать ]Трансформация ФЛ в более агрессивное состояние или другой тип агрессивной лимфомы связана с: 1) преимущественно мутациями, активирующими гены CREEBP, KMT2D, STAT6, CARD11 (кодирующими гуанилаткиназу , которая взаимодействует с BCL10 и активирует NF-κB для регуляции выживание клеток); 2) изменения в экспрессии разнообразных генов; 3) перепроизводство различных цитокинов , активирующих клетки [25] и CD79B (кодирующий белковый компонент Ig-бета рецептора B-клеток). [26] ); 4) ген-инактивирующие мутации в TNFAIP3, CD58 (кодирующий молекулу клеточной адгезии , антиген 3, связанный с функцией лимфоцитов, который участвует в активации Т-клеток). [27] ), CDKN2A (кодирующий p16INK4a и p14arf супрессоры опухолей белки- [28] ) или CDKN2B (кодирующий ингибитор циклинзависимой киназы 2B, множественный супрессор опухоли 2 [29] ) (инактивация любого гена CDKN2 вызывает нестабильность генома , т.е. увеличение частоты мутаций других генов) и TNFRSF4 (кодирующий один тип рецептора фактора некроза опухоли). [30] ); и 5) мутации, активирующие или инактивирующие ген, или другие причины недостаточной или сверхэкспрессии c-MYC ((кодирующего протоонкогена фактор транскрипции c-Myc , который регулирует экспрессию разнообразных генов, многие из которых способствуют пролиферации клеток [31] ). [24]
Окружающая среда опухоли
[ редактировать ]Неопухолевые иммунные и стромальные клетки , а также внеклеточный матрикс в тканях могут позволить неопластическим фолликулярным клеткам выживать, пролиферировать и избегать наблюдения со стороны иммунной системы . Например, лабораторные исследования показывают, что: 1) фолликулярные дендритные клетки , фибробластические ретикулярные клетки и Т-хелперные клетки передают сигналы роста и выживания неопластическим фолликулярным В-клеткам; 2) неопластические фолликулярные В-клетки рекрутируют регуляторные Т-клетки , которые подавляют иммунный ответ на них; 3) цитотоксические Т-клетки , которые обычно убивают неопластические клетки, становятся дисфункциональными в присутствии неопластических фолликулярных клеток, встроенных в эту многоклеточную среду; и 4) костного мозга стромальные клетки непосредственно поддерживают рост неопластических фолликулярных клеток. [24] Было показано, что снижение уровня иммунной инфильтрации тесно связано с ранним прогрессированием заболевания. [32]
Презентация и курс
[ редактировать ]Фолликулярная лимфома in situ
[ редактировать ]ФЛ обычно предшествует, но редко прогрессирует до ИСФЛ, бессимптомного заболевания, которое обычно обнаруживается в тканях, биопсированных по другим причинам. Лимфома ФЛ может быть диагностирована в редких случаях, когда у людей с ИСФЛ при последующих обследованиях обнаруживается ФЛ. [9] Аналогичным образом, люди с >1 из 10 000 циркулирующих лимфоцитов, содержащих транслокацию t(14:18)q32:q21), имеют повышенный, но все же небольшой риск развития ФЛ и постановки диагноза ФЛ при последующих обследованиях. [10]
Фолликулярная лимфома
[ редактировать ]ФЛ обычно проявляется бессимптомным увеличением лимфатических узлов на шее, подмышках, в паху, [13] бедренный канал , [33] или других локализациях у лиц (средний возраст 65 лет) без известного анамнеза ИСФЛ или аномального количества циркулирующих t(14:18)q32:q21-конатианизирующих лимфоцитов. [13] Эти увеличения могли присутствовать от месяцев до лет и за это время то увеличиваться, то уменьшаться в размерах. [8] Реже ФЛ проявляется в виде экстраузловых образований на коже, щитовидной железе, слюнной железе, молочной железе, яичках. [11] селезенка , печень, [33] и/или легкое. [4] Независимо от типа проявления ФЛ обычно (~80% случаев) [8] ) на поздней стадии диагностики, о чем свидетельствует поражение костного мозга (50% [13] до 70% [8] случаев), множественные лимфатические узлы в разных частях тела, [9] и/или другие ткани. [11] Меньшинство (<33%) [8] у пациентов с ФЛ наблюдаются симптомы В , т.е. рецидивирующая необъяснимая лихорадка , рецидивирующая ночная потливость и/или потеря веса ≥10% за последние 6 месяцев. [5] Как правило, заболевание имеет вялотекущее и продолжительное течение со средней продолжительностью жизни 15–20 лет: большой процент пациентов умирают от других причин, отличных от заболевания ФЛ. [5] Однако каждый год, включая первые годы после постановки диагноза, около 2–3% случаев ФЛ трансформируются в т-ФЛ; [12] Медиана выживаемости составила ~4,5 года после начала этой трансформации. [5]
Существуют менее распространенные подтипы ФЛ, которые различаются не только по своей картине, но и по гистопатологии , генетическим аномалиям и течению. Этими подтипами, которые в настоящее время (т.е. первичная ФЛ желудочно-кишечного тракта) или могут в будущем (ФЛ детского типа) считаться самостоятельными заболеваниями, являются:
Фолликулярная лимфома двенадцатиперстной кишки
[ редактировать ]Фолликулярная лимфома двенадцатиперстной кишки (ДФЛ) первоначально считалась разновидностью первичной фолликулярной лимфомы желудочно-кишечного тракта (ЖКТ) (ПГТФЛ), т.е. фолликулярной лимфомы, при которой поражения ЖКТ были заметной частью заболевания. [34] Однако в некоторых случаях PGTFL поражения локализовались в двенадцатиперстной кишке и других частях тонкой кишки , обычно без вовлечения других частей желудочно-кишечного тракта или тканей за пределами желудочно-кишечного тракта. Это контрастирует с другими случаями PGTFL, которые представляли собой системные заболевания, поражающие широкий спектр тканей желудочно-кишечного тракта и тканей, не относящихся к желудочно-кишечному тракту. Следовательно, Всемирная организация здравоохранения (2017) исключила локализованное заболевание из категории первичных фолликулярных лимфом желудочно-кишечного тракта, реклассифицировала его как отдельную болезнь и назвала фолликулярной лимфомой двенадцатиперстной кишки. [6] ДФЛ чаще всего является бессимптомным заболеванием, которое диагностируется при эндоскопическом исследовании ЖКТ, проводимом по другим причинам. Реже он проявляется неясными абдоминальными симптомами. [35] [36] В одном обзоре предыдущих исследований очаги поражения в 85% случаев первичной фолликулярной лимфомы двенадцатиперстной кишки локализовались не только в двенадцатиперстной кишке, но и в других участках кишечника (например, тощей и/или подвздошной кишке ). [11] в редких случаях наблюдаются поражения прямой кишки [37] или слепая кишка [38] PDF — это вялотекущее заболевание, которое может спонтанно регрессировать и рецидивировать, но лишь в редких случаях прогрессирует до более агрессивной формы. Стратегия «наблюдай и жди» обычно рекомендуется для начального лечения заболевания. [39]
Первичная фолликулярная лимфома желудочно-кишечного тракта
[ редактировать ]PGTFL — это фолликулярная лимфома (которая, согласно нынешнему определению, исключает случаи фолликулярной лимфомы двенадцатиперстной кишки), которая имеет выраженный компонент поражения желудочно-кишечного тракта. Заболевание может проявляться признаками и симптомами, типичными для обычного типа фолликулярной лимфомы. Например, увеличение лимфатических узлов на шее, подмышках, в паху, [13] бедренный канал и/или другие области, [33] и/или признаки и симптомы заболевания желудочно-кишечного тракта [34] вследствие поражения желудка, тонкой кишки, толстой кишки [11] или прямая кишка может быть видна. [37] Эти признаки и симптомы могут включать боль в животе, непроходимость кишечника , [11] постоянная тошнота и рвота, гематохезия (т. е. выделение свежей крови, обычно с фекалиями через прямую кишку) или мелена (т. е. выделение смолистых фекалий, содержащих кровь, переваренную в желудке или верхних отделах кишечника). [40] PGTFL обычно лечат так же, как и случаи обычной фолликулярной лимфомы: в зависимости от тяжести заболевания и его симптомов пациентов лечат с бдительным ожиданием , хирургическим вмешательством, химиотерапией, лучевой терапией, иммунотерапией плюс лучевая терапия или комбинацией этих методов. [40]
Преимущественно диффузная фолликулярная лимфома с делецией 1p36.
[ редактировать ]Преимущественно диффузная фолликулярная лимфома с делецией 1p36 является редким подтипом ФЛ. [7] при котором в пораженных лимфатических узлах наблюдаются инфильтрации центроцитов и центобластов, которые обычно не образуют узловатых, закрученных узоров, характерных для большинства типов ФЛ. [1] Кроме того, в этих клетках отсутствует транслокация t(14:18)(q32:q21.3), обычно встречающаяся при других типах ФЛ, но, как и во многих случаях ФЛ, имеется делеция в терминальной части короткого (т.е. «p» ) плечо хромосомы 1, кодирующее ген TNFRSF14 (см. раздел патофизиологии). [13] Преимущественно диффузная фолликулярная лимфома с делецией 1p36 обычно проявляется массивным увеличением паховых (т. е. паховых) лимфатических узлов , но может проявляться увеличением подмышечных (т. е. подмышечных) или шейных (т. е. шеи) лимфатических узлов . В редких случаях возможно поражение костного мозга . Несмотря на наличие обширного и диссеминированного заболевания, преимущественно диффузная фолликулярная лимфома с делецией 1p36, по-видимому, является вялотекущим заболеванием, которое может потребовать длительного наблюдения, а не чрезмерного лечения. [7]
Фолликулярная лимфома детского типа
[ редактировать ]Первоначально сообщалось, что фолликулярная лимфома детского типа (ПТФЛ) возникает у детей в возрасте 1–17 лет (средний возраст ~ 13–14 лет), но позже сообщалось, что она возникает и у взрослых. [41] Это расстройство недавно было определено Всемирной организацией здравоохранения (2016 г.) как отдельное заболевание, которое встречается преимущественно у мужчин. [7] и поражает опухшие лимфатические узлы головы (включая миндалины и аденоиды ), шеи, [41] или, реже, в подмышечных, паховых областях или нелимфоидных тканях. [42] Однако в настоящее время у пациентов, у которых наблюдалось или наблюдается поражение областей или тканей за пределами головы, шеи, подмышек или паха, теперь считается гораздо более вероятным наличие нового и предварительно определенного заболевания — крупноклеточной В-клеточной лимфомы с Перегруппировка IRF4 . [41]
Поражения при ПТФЛ состоят из инфильтратов, содержащих быстро пролиферирующие центроциты и центробласты, лишенные транслокации t(14:18)(q32:q21.3), но, тем не менее, часто сверхэкспрессирующие ген BCL2 . [7] Эти клетки могут демонстрировать потерю гетерозиготности по 1p36 (20–50% случаев), что приводит к снижению экспрессии гена TNFRSF14 (см. раздел «Патофизиология»), а также к мутациям в IRF8 (10–50% случаев), что способствует на развитие и функционирование В-клеток, [43] [44] и ген MAP2K1 (10–40% случаев), который регулирует активацию сигнального пути клеток ERK. [45] Сообщается, что более 2 дюжин других генов мутируют в редких случаях ПТФЛ, но в целом генетические аномалии, обнаруживаемые при этом заболевании, меньше и менее сложны, чем таковые при других типах ФЛ. [42] ПТФЛ имеет вялотекущее, рецидивирующее и ремиттирующее течение с 5-летней выживаемостью >95%. [42] Пациентов с диагнозом ПТФЛ лечили химиотерапией, хирургическим вмешательством и комбинацией этих методов лечения. В целом у этих пациентов дела шли хорошо (100% выживаемость при <5% случаев рецидивов независимо от метода лечения). Совсем недавно 36 пациентам была проведена только хирургическая резекция с последующим наблюдением; все эти пациенты выжили, и только у одного случился рецидив. Таким образом, ПТФЛ, по-видимому, является крайне вялотекущим типом ФЛ, при котором многочисленные исследования сообщают об общей выживаемости и выживаемости без прогрессирования 100% и >90% соответственно в течение >2 лет и предполагаемой вероятности 5-летнего отсутствия событий. выживаемость ~96%. Терапевтические схемы и последующие наблюдения, которые лучше всего лечат это расстройство у детей, подростков и взрослых (взрослым может потребоваться другое лечение, чем детям и подросткам), требуют дальнейшего изучения. [41]
Первичная фолликулярная лимфома яичка
[ редактировать ]Первичная фолликулярная лимфома яичка (ПФЛТ), также называемая фолликулярной лимфомой яичка , была классифицирована Всемирной организацией здравоохранения как отдельная форма ФЛ в 2016 году. [33] Это чрезвычайно редкое заболевание, которое встречается преимущественно у детей и подростков. [46] но также сообщалось у 5 взрослых. [47] PFLT отличается от случаев типичной фолликулярной лимфомы, поражающей яички, тем, что чаще встречается у детей и подростков; вовлекаются злокачественные B-клетки, имеющие транслокацию t(14:18)q32:q21); и проявляется заболеванием, которое строго ограничено яичками. Хотя PFLT похож на фолликулярную лимфому детского типа тем, что не затрагивает клетки, несущие транслокацию t(14:18)q32:q21), он отличается от первого заболевания тем, что он ограничен яичками и включает злокачественные клетки, не экспрессирующие Bcl2. . [48] ПФТЛ представляет собой крайне вялотекущее заболевание, которое проявляется поражениями, имеющими типичную гистологию ФЛ или, чаще, смешанную гистологию ФЛ-диффузной крупноклеточной лимфомы. Обычно это поражение одного яичка размером 2–4 сантиметра. Лечение пациентов включало удаление пораженных яичек с последующим применением различных стандартных схем противолимфомной химиотерапии для достижения отличных результатов, т.е. 100% завершенной ремиссии без рецидивов заболевания у 15 пациентов детей и подростков, наблюдаемых в течение 4–96 месяцев. Не зарегистрировано ни одного случая прогрессирования первичной фолликулярной лимфомы яичка в Т-ФЛ. Хирургическое вмешательство с последующей менее интенсивной химиотерапией или даже без нее может оказаться оптимальным лечением этого заболевания. [46]
Трансформированная фолликулярная лимфома
[ редактировать ]ФЛ прогрессирует со скоростью 2–3% в год, по крайней мере, в течение первых 10 лет после постановки диагноза, переходя в более агрессивную форму, главным образом диффузную крупноклеточную В-клеточную лимфому (~93% случаев) или подобную лимфому Беркитта (~7% случаев). случаев) или в редких случаях имеют гистологию, напоминающую предшественник В-клеточного лимфобластного лейкоза , плазмобластную лимфому , подтип В-клеточной лимфомы высокой степени злокачественности , лимфому Ходжкина В-клеточного типа, хронический лимфоцитарный лейкоз/мелкоклеточную лимфоцитарную лимфому , [5] или гистиоцитарная саркома . [1] Т-ФЛ почти всегда диагностируется у пациентов, находящихся под наблюдением по поводу ФЛ. У этих пациентов с ФЛ наблюдаются: быстрый рост лимфатических узлов; образование экстраузловых поражений в экстраузловых участках, таких как центральная нервная система , печень или кости; появление B-симптомов (т.е. лихорадки, ночной потливости , потери веса); развитие гиперкальциемии (т.е. высоких уровней кальция в сыворотке); и/или внезапное повышение уровня фермента лактатдегидрогеназы в сыворотке крови . [5] Меньшая часть пациентов с Т-ФЛ не имеет в анамнезе ФЛ. У этих пациентов обычно наблюдается распространенное, обширное заболевание, которое может сопровождаться экстраузловыми поражениями и B-симптомами. [1] Как правило, все различные формы Т-ФЛ представляют собой агрессивные, быстро прогрессирующие заболевания с общим временем выживания в среде у пролеченных пациентов ~4,5 года. [1] [5] Трансформация FL в DLBCL более чем в 70% случаев связана с усилением активности MYC за счет генетических или негенетических механизмов. [49]
Диагностика
[ редактировать ]
Диагноз ФЛ зависит от исследования пораженных тканей на наличие гистологических , иммунологических и хромосомных аномалий, которые указывают на заболевание. ФЛ обычно поражает увеличенные лимфатические узлы, заполненные аномальными фолликулами (см. рисунок рядом), которые при гистологическом исследовании содержат смесь центроцитов или центробластов, окруженных доброкачественными клетками, в основном Т-клетками . Центроциты, число которых обычно превышает число центробластов, представляют собой В-клеточные лимфоциты малого и среднего размера, для которых характерно расщепленное ядро ; центропбласты представляют собой более крупные В-клеточные лимфоциты без расщепленных ядер. [11] В редких случаях ФЛ могут наблюдаться поражения, которые содержат тканевые инфильтраты, в которых преобладают В-клетки с признаками клеток-предшественников (т.е. «бластных») , моноцитов или злокачественных клеток мантии, таких как те, которые обнаруживаются при лимфоме из клеток мантийной зоны . [1] Иммунохимический анализ показывает, что эти клетки обычно экспрессируют поверхностные маркеры В-клеток, включая CD10 (60% случаев), CD20 , CD19 , CD22 и CD79 , но не CD5 , CD11c или CD23 ; белки клеточной поверхности [4] геномный анализ показывает, что эти клетки содержат транслокацию t(14:18)(q32:q21.3) (85–90% случаев), делеции 1p36 (60–70% случаев) и с гораздо меньшей частотой другие геномные аномалии. перечислены в разделах «Патофизиология», «Презентация» и «Курс» выше. Ни один из этих белковых маркеров или геномных аномалий не является диагностическим для ФЛ, например, транслокация t(14:18)(q32:q21.3) обнаруживается в 30% случаев диффузной крупноклеточной В-клеточной лимфомы и в небольшом количестве реактивных доброкачественных лимфом. узлы. Скорее, диагноз ставится на основании сочетания гистологических, иммунологических и геномных отклонений. [4] Согласно критериям Всемирной организации здравоохранения (ВОЗ), фолликулярную лимфому можно классифицировать морфологически по относительному количеству центробластов . Однако такая классификация необязательна из-за плохой воспроизводимости и небольшой разницы в прогнозе и лечении, за исключением того, что лимфому, содержащую почти только центробласты, можно диагностировать как диффузную крупноклеточную В-клеточную лимфому (ДКБКЛ). [50] Факультативная классификация фолликулярной лимфомы следующая: [51]

- Центроциты от малого до среднего размера с угловатыми, удлиненными, расщепленными или скрученными ядрами.
- Центробласты – это более крупные клетки, содержащие везикулярные ядра с одним-тремя базофильными ядрышками, прилегающими к ядерной мембране.
- Фолликулярные дендритные клетки имеют округлые ядра, центрально расположенные ядрышки, мягкий и рассеянный хроматин и уплощенную прилежащую ядерную мембрану.
- Степень 1: фолликулы содержат <5 центробластов в поле зрения под большим увеличением (hpf).
- Степень 2: фолликулы имеют от 6 до 15 центробластов на один hpf.
- Степень 3: фолликулы содержат >15 центробластов на один час после выздоровления.
- Степень 3А: Степень 3, при которой фолликулы содержат преимущественно центроциты.
- Степень 3B: Степень 3, при которой фолликулы почти полностью состоят из центробластов.
1 и 2 степени считаются FL низкой степени; Степень 3А обычно также считается ФЛ низкой степени, хотя в некоторых исследованиях ее рассматривают как ФЛ высокой степени; а степень 3B считается очень агрессивным FL в категории t-FL. [8]
В дополнение к заболеванию 3В степени гистологическое исследование может выявить другие признаки Т-ФЛ, такие как гистологические данные, соответствующие ФЛ и диффузной крупноклеточной лимфоме в той же ткани (называемой составной лимфомой ) или в отдельных тканях (называемой ( дискордантная лимфома) лимфомы ) или гистологические данные, сходные с таковыми при лимфоме Беркитта, В-клеточном лимфобластном лейкозе-предшественнике, плазмобластной лимфоме, подтипе В-клеточной лимфомы высокой степени злокачественности, лимфоме Ходжкина В-клеточного типа, хроническом лимфоцитарном лейкозе/мелкоклеточной лимфоцитарной лимфоме , [5] или гистиоцитарная саркома. [1] Другие данные, указывающие на наличие этой трансформации, включают быстрый рост размеров лимфатических узлов, недавно приобретенные или новые симптомы B , недавнее развитие поражений FL в неузловых тканях, быстрое повышение уровня лактатдегидрогеназы в сыворотке и наличие высоких уровней сывороточный кальций . [12]
Дифференциальный диагноз
[ редактировать ]
ФЛ можно спутать с В-клеточной лимфомой маргинальной зоны , лимфомой мантийных клеток и вариантом мелкой лимфоцитарной лимфомы хронического лимфоцитарного лейкоза . Злокачественные клетки В-клеточной лимфомы маргинальной зоны могут образовывать фолликулярные структуры, но обычно пролиферируют в маргинальной зоне, а не в зародышевом центре лимфоидной ткани. Эти злокачественные клетки часто имеют черты моноцитов или плазматических клеток . В мантийно-клеточных лимфомах наблюдаются монотонные лимфоциты среднего размера, моноциты и атрофированные зародышевые центры; в отличие от ФЛ, злокачественные лимфоциты при этом заболевании положительны на циклин D1 при иммуногистохимическом окрашивании . Малые лимфоцитарные лимфомы состоят из узловых структур со злокачественными клетками малого и среднего размера, окружающими незрелые лимфоциты и иммунобласты . Злокачественные клетки при этом заболевании, в отличие от ФЛ, окрашиваются положительно на CD5 и CD23 . [11]
Лечение и прогноз
[ редактировать ]ФЛ обычно представляет собой медленно растущую лимфому с общей средней продолжительностью жизни пролеченных пациентов 10–15 лет. [34] во многих случаях размер поражений увеличивается и уменьшается, а в редких случаях спонтанно прекращается. [4] Эти соображения отдают предпочтение использованию наблюдения перед вмешательством у пациентов, у которых конкретная форма ФЛ имеет благоприятный прогноз или у которых непереносимость агрессивного лечения. [4] Однако большинство случаев ФЛ имеют менее благоприятный прогноз на определенной стадии заболевания и поэтому требуют вмешательства. Существует мало консенсуса относительно рекомендаций, заболевания которые следует использовать для определения прогноза и лечения ФЛ при ее проявлении или во время ее течения. В настоящее время используемые для этого индикаторы включают: 1) гистологию ; 2) подтип; 3) предсказанная праздность и потенциал трансформации; и 4) степень заболевания, измеренная с помощью клинических обследований, биопсии костного мозга для определения поражения костного мозга и ПЭТ/КТ грудной клетки, живота, таза и любых областей за пределами этих областей, если физическое обследование предполагает поражение. [52] Некоторые предлагаемые рекомендации по использованию этих параметров для определения прогноза и необходимости лечения ФЛ включают: [8]
- Критерии ВОЗ с использованием гистологической степени (см. предыдущий раздел): У пациентов со степенью заболевания 1, 2 и 3А прогнозируется такой же низкий прогноз риска, который наблюдается в случаях типичной ФЛ, в то время как у пациентов со степенью заболевания 3В прогнозируется наличие прогноз высокого риска типичен для Т-ФЛ.
- ( Международный прогностический индекс фолликулярной лимфомы FLIPI): FLIPI использует следующие критерии: возраст ≥60 лет; Болезнь Анн-Арбор, стадия III (т. е. поражения, расположенные как выше, так и ниже грудной диафрагмы ) или IV (т. е. диссеминированные поражения, затрагивающие один или несколько нелимфатических органов); гемоглобин крови <12 грамм/децилитр; уровень лактозодегидрогеназы в сыворотке крови выше нормы; и поражение >4 лимфатических узлов. Пациенты с положительным результатом по 0–1, 2 или ≥3 из этих факторов классифицируются как группы низкого, среднего и высокого риска соответственно, и после лечения схемами, включающими ритуксимаб, прогнозируемые показатели выживаемости без прогрессирования в течение 2 лет составляют 84, 72 и 65% соответственно, а 5-летняя выживаемость составила 98, 94 и 87% соответственно. [4]
- Индекс FLIP2. В этой модификации FLIP1 используется возраст ≥60 лет; гемоглобин крови <12 грамм/децилитр; уровень лактозодегидрогеназы в сыворотке выше нормы; уровень бета-2-микроглобулина в сыворотке выше нормы; ≥1 лимфатический узел диаметром >6 сантиметров; и вовлечение костного мозга. Прогнозируемый процент пациентов, получавших терапию, с выживаемостью без прогрессирования в течение 5 лет для лиц с положительным результатом по 0, 1–2 и ≥3 из этих факторов составляет 80, 51 и 19% соответственно. [8]
- КТ/ПЭТ-визуализация: этот метод измеряет общий объем опухоли тела, определяемый по поглощению тканями радиоактивной флюдезоксиглюкозы (F 18 ). Сообщается, что без прогрессирования и общая выживаемость в течение 5 лет для пациентов с предполагаемым объемом опухоли выше и ниже 510 кубических сантиметров составляет 32,7 и 84,8% против 65,1 и 94,7% соответственно. [8]
- Стадия Лугано: этот метод классифицирует стадию I как поражение одной лимфатической области или внелимфатического участка; Стадия II заболевания с поражением ≥2 лимфатических участков или 1 лимфатического узла плюс 1 экстралимпатический участок, при этом все поражения находятся на одной стороне диафрагмы; Заболевание стадии III с поражением ≥2 лимфатических областей, расположенных на противоположных сторонах диафрагмы; и стадия IV заболевания в виде диссеминированных поражений, которые обнаруживаются в ≥1 нелимфатическом органе. [4]
- Прогноз, основанный на ответе: прогнозируется, что у пациентов с ФЛ, у которых заболевание прогрессирует в течение 24 месяцев после начала лечения химиотерапией и иммунотерапией, по сравнению с пациентами, у которых заболевание не прогрессирует в течение 24 месяцев, 5-летняя выживаемость составит 50–74% против ~90% соответственно. [8]
Прогноз и лечение специфических проявлений типичных случаев ФЛ (прогнозы и рекомендации по лечению первичной ФЛ желудочно-кишечного тракта, преимущественно диффузной ФЛ с делецией 1p36, ФЛ педиатрического типа и первичной ФЛ яичка см. выше) обычное использование следующее:
Фолликулярная лимфома in situ
[ редактировать ]ИСФЛ — доброкачественное состояние, которое можно периодически обследовать, чтобы выявить редкие случаи, которые переходят в ФЛ; в противном случае ИСФЛ не лечится. [9]
Локализованная фолликулярная лимфома
[ редактировать ]В 10–20% случаев ФЛ ограничивается одним полем облучения, не затрагивает костный мозг и поэтому расценивается как локализованная ФЛ ранней стадии. В этих случаях, которые иногда классифицируются как стадия I по Анн-Арбору (т. е. заболевание, ограниченное одной ограниченной областью) или стадия II (т. е. заболевание, ограниченное двумя участками, расположенными по одну сторону диафрагмы), [4] лучевая терапия обеспечивает общую 10-летнюю выживаемость 60–80% и медианную общую выживаемость 19 лет. [8] Кажется вероятным, что многие из рецидивов в этих случаях обусловлены невыявленным заболеванием за пределами поля облучения во время лучевого лечения. Настоятельно рекомендуется использовать ПЭТ/КТ, чтобы убедиться в локализации ФЛ. В любом случае, отличные результаты, достигнутые с помощью лучевой терапии, убедительно подтверждают ее использование при локализованных заболеваниях. Использование иммунотерапевтического агента , такого как ритуксимаб, отдельно или в сочетании с химиотерапевтическим режимом, таким как CVP (т.е. циклофосфамид , винкристин , преднизолон и ритуксимаб ), в случаях локализованного заболевания на ранней стадии может быть подходящим выбором для некоторых из этих ранних стадий. пациенты. [4] Однако последний подход рекомендуется для случаев локализованного заболевания, при котором заболевание выходит за пределы одного поля: 56% пациентов, получавших такое лечение, имели выживаемость без прогрессирования в течение 10 лет, в то время как у пациентов, получавших другие схемы, выживаемость без прогрессирования составила 41 %. Тем не менее, общая выживаемость не различалась между двумя группами. [13]
Бессимптомная фолликулярная лимфома
[ редактировать ]Пациенты с бессимптомной, но не локализованной ФЛ низкой степени тяжести, [8] [53] [54] желудочно-кишечный тракт ФЛ, [34] и фолликулярная лимфома детского типа [41] прошли тщательное наблюдение без терапевтического вмешательства. Даже агрессивная, рецидивирующая или трансформированная ФЛ высокой степени тяжести может также наблюдаться у бессимптомных пациентов. У бессимптомных пациентов, которым было рекомендовано в качестве триггера для начала лечения, относятся один или несколько из следующих признаков: размер опухоли ≥7 см в диаметре; вовлечение ≥3 узлов в 3 отдельных областях, каждый из которых имеет диаметр ≥3 см; сдавление органа; наличие асцита или плеврального выпота (т.е. скопление жидкости в брюшной или плевральной полостях); плохой работоспособный статус из-за заболевания; повышенный уровень сывороточной лактозодегидрогеназы или бета-2-микроглобулина ; [4] наличие локализованных поражений костей; поражение почек; снижение уровня циркулирующих тромбоцитов или любого из различных типов лейкоцитов ; появление значительного зуда (т.е. ощущения зуда) или других симптомов группы B; и увеличение (т.е. увеличение размера на ≥50% в течение периода не менее 6 месяцев) лимфатических узлов, селезенки или других органов или тканей, инфильтрированных фолликулярной лимфомой. [33]
Симптоматическая фолликулярная лимфома
[ редактировать ]Симптоматическая ФЛ требует лечения, направленного на облегчение симптомов за счет снижения нагрузки опухолевых клеток. различные химиотерапевтические Для этого использовались схемы, включая комбинации алкилирующих противоопухолевых агентов , аналогов нуклеозидов и/или антрациклинов . Двумя обычно используемыми химиотерапевтическими режимами являются CVP (см. раздел «Локализованная ФЛ») и CHOP (т. е. CVP плюс антрациклин адриамицин ). Новые препараты, используемые для лечения ФЛ, включают моноклональные антитела, такие как ритуксимаб , обинутузумаб , галиксимаб , инотузумаб озогамицин , или эпратузумаб а также иммуномодуляторы, такие как леналидомид и интерферон . Последние препараты использовались в комбинации или по отдельности для лечения симптоматической ФЛ. [13] В большинство таких схем добавляются ритуксимаб (моноклональное антитело, которое связывает и тем самым убивает белок клеточной поверхности CD20 на В-клетках) с режимами CVP или CHOP (называемыми режимами R-CVP и R-CHOP).
Схема R-CHOP, по-видимому, превосходит схему R-CVP: например, в одном исследовании было обнаружено, что 8-летняя выживаемость без прогрессирования составила 57% против 46% для двух соответствующих схем. [33] Совсем недавно пациентов с ФЛ лечили другими схемами, включая: 1) ритуксимаб в сочетании с химиотерапевтическим алкилирующим агентом бендамустином ; 2) в сочетании с химиотерапевтическим средством флударабином и ингибитором топоизомеразы II типа ; митоксантроном ритуксимаб [33] и 3) ритуксимаб в сочетании с другим иммунотерапевтическим агентом, таким как галиксимаб , эпратузумаб соответственно против белков клеточной поверхности CD80 или CD22 на иммунных клетках, включая В-клетки), или иммуномодулирующий препарат леналидомид (моноклональные антитела, направленные . [13] Хотя еще слишком рано судить о долгосрочных результатах последних схем, эти схемы показали схожие результаты при анализе на основании плохих ответов на лечение (~ 10–20% плохих ответов). Бендамустин в сочетании с ритуксимабом может быть предпочтительнее R-CHOP или R-CVP для лечения ФЛ низкой степени тяжести (т. е. степени 1, 2 и, возможно, 3А); R-CHOP может быть предпочтителен при ФЛ, которая имеет характеристики высокого риска (например, высокий уровень макроглобулина бета-2 или поражение костного мозга). Комбинация леналидомида с ритуксимабом показала хороший потенциал в лечении вялотекущих случаев ФЛ. [13]
Исследования показывают, что поддерживающая терапия ритуксимабом после успешной индукционной терапии продлевает выживаемость без прогрессирования заболевания; например, одно исследование показало, что выживаемость без прогрессирования после 6 лет лечения составила 59,2% у пациентов, получавших поддерживающую терапию ритуксимабом, и 42,7% без этой поддерживающей терапии; однако общая выживаемость через 6 лет была одинаковой в обеих группах: 87,4% и 88,7% соответственно. Другое исследование показало, что длительное лечение ритуксимабом не принесло никаких преимуществ в течение восьмимесячного периода лечения. [13] Наконец, операция [55] [56] и радиация [4] [13] [33] представляют собой дополнительные методы лечения, которые можно использовать для облегчения симптомов, вызванных объемным заболеванием t-FL, или для лечения поражений у пациентов, которые не могут выдержать другие виды лечения.
Трансформированная фолликулярная лимфома
[ редактировать ]Ранние исследования по лечению Т-ФЛ различными схемами чисто химиотерапии дали плохие результаты со средней общей выживаемостью 1–2 года. Однако добавление ритуксимаба к таким схемам, как CVP и CHOP, в рамках индукционной и поддерживающей терапии (т.е. R-CVP и R-CHOP) значительно улучшило общую 5-летнюю выживаемость до 73%. Режим R-CHOP является хорошим вариантом лечения таких случаев. [5] Однако эти схемы не обязательно начинать у людей с бессимптомной ФЛ и низкой опухолевой нагрузкой: результаты у таких пациентов не показывают разницы между ранним и отсроченным лечением. Некоторые недавние исследования показали, что использование ритуксимаба в сочетании с бендамустином (т.е. схема RB) дает лучшие результаты, чем R-CHOP: время выживаемости без прогрессирования в одном исследовании составило 69,5 месяцев для RB и 31,2 месяца для R-CHOP. Аналогичные результаты были получены при сравнении RB с R-CVP. Эти исследования также не выявили общего улучшения времени выживания между схемами RB и R-CHOP. Другие недавно рассмотренные схемы включают 1) использование обинутузумаба вместо ритуксимаба в группах R-CHOP и R-CVP для достижения показателей выживаемости без прогрессирования в течение 3 лет, равных 80% для режима химиотерапии с обинутузумабом против 73% для режима ритуксимаба. режим химиотерапии и 2) комбинация ритуксимаба с леналидомидом (без химиотерапевтического агента) в сравнении с различными видами химиотерапии и иммунотерапией (в основном ритуксимабом) для достижения аналогичных показателей полной ремиссии и 3-летней выживаемости без прогрессирования, но сочетание ритуксимаба с леналидомидом вызывает меньшую токсичность (т.е. тяжелую токсичность). нейтропения ). Во многих из этих исследований после индукционной терапии применялась поддерживающая терапия ритуксимабом. [4]
Профилактика
[ редактировать ]Некоторые исследования, хотя и не окончательные, предполагают, что раннее лечение ФЛ низкого риска снижает частоту прогрессирования заболевания в Т-ФЛ. Лечение, используемое в этих исследованиях, включает химиотерапию, лучевую терапию и комбинации иммунотерапии плюс поддерживающую терапию ритуксимабом. [12]
Рецидивирующая фолликулярная лимфома
[ редактировать ]Пациенты, у которых наблюдается рецидив после начальной терапии ФЛ, могут находиться под тщательным наблюдением без терапии, если симптомы отсутствуют. При необходимости лечения пациентов можно лечить по первоначальной схеме лечения, если такое лечение привело к ремиссии, продолжавшейся не менее одного года; в противном случае используется альтернативный режим. [13] Схемы, обычно используемые при рецидиве лимфомы, включают R-CHOP, R-CVP, RFM (т.е. ритуксимаб, флударабин и митоксантрон ) и RB (бендамустин плюс ритуксимаб). [4] Пациенты, у которых на ранней стадии лечения (например, в течение 1–2 лет после первоначального лечения) или с множественными рецидивами, также лечились аутологичными ( т. е. стволовыми клетками, взятыми у пациента), или аллогенными (т. е. стволовыми клетками, взятыми от донора) стволовыми клетками костного мозга. трансплантация. Хотя исследования не дают окончательных результатов, трансплантация костного мозга аутологичных стволовых клеток, по-видимому, продлевает выживаемость у пациентов с ранней неудачей лечения, которые достаточно здоровы, чтобы выдержать эту терапию. Пациенты, непригодные для лечения, могут получить пользу от первоначального лечения обинутузумабом в сочетании с бендамустином с последующим поддерживающим лечением обинутузумабом (если они ранее не получали лечение обинутузумабом). [13]
Другие в основном экспериментальные методы лечения, которые в настоящее время изучаются у пациентов с множественными неудачами лечения, включают: 1) Ингибиторы фосфоинозитид-3-киназы, такие как копанлисиб , дувелисиб и иделалисиб , которые блокируют сигнальный путь фосфоинозитид-3-киназы , который способствует выживанию, пролиферации и другим потенциально злокачественным заболеваниям. поведение клеток; 2) инфузия тисагенлеклейцела Т-клеток химерного антигенного рецептора (т.е. Т-клеток CAR) (т.е. Т-клеток, которые были выделены от пациентов и сконструированы для экспрессии рецептора белка CD19 на Т-клетках и, таким образом, их уничтожения, а затем введены обратно в пациент-донор); [52] 3) ингибитор тирозинкиназы Брюона , ибрутиниб , для блокирования действия этой кианазы на созревание В-клеток; 4) ингибитор BCL венетоклакс для блокирования действия Bcl2, способствующего выживанию и пролиферации B-клеток; 5) ингибиторы гистондеацетилазы абексиностат и таземетостат для модификации экспрессии различных генов; и 6) Ингибиторы контрольных точек ниволумаб , пидилизумаб и пембролизумаб, способствующие способности иммунной системы подавлять рост раковых клеток. [4] В предварительных исследованиях на пациентах с ФЛ, которые были известны или считались невосприимчивыми к более традиционным методам лечения, эти препараты в сочетании с более традиционными препаратами, особенно ритуксимабом, дали многообещающие результаты. Ингибиторы фосфоионитид-3-киназы дали общий ответ на лечение в течение 10–12,5 месяцев у 42–59%; Клетки tisagenlecleuce дали общий уровень ответа без прогрессирования в 70% после 28-месячного наблюдения; [52] Ингибиторы фосфоинозитид-3-киназы давали общий уровень ответа ~40% и полный ответ 1–20%; Ингибитор тирозинкиназы Брутона дал общий и полный ответ на лечение в 38% и 18% соответственно; ингибитор Bcl дает общий и полный ответ в 33% и 14% соответственно; ингибиторы гистондеацетилазы дают общий уровень ответа 35–71%; и ингибиторы контрольных точек дают общий уровень ответа 40–80% и полный ответ 10–60%. [4]
См. также
[ редактировать ]- in situ Фолликулярная лимфома
- Крупноклеточная лимфома
- Список гематологических заболеваний
- Тоцилизумаб
Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д и ж г час Ксерри Л., Дирнхофер С., Кинтанилья-Мартинес Л., Сандер Б., Чан Дж.К., Кампо Е. и др. (февраль 2016 г.). «Гетерогенность фолликулярных лимфом: от раннего развития к трансформации». Архив Вирхова . 468 (2): 127–39. дои : 10.1007/s00428-015-1864-y . ПМИД 26481245 . S2CID 2978889 .
- ^ «фолликулярная лимфома» в Медицинском словаре Дорланда.
- ^ Крупноклеточная + лимфома + фолликулярная в Национальной медицинской библиотеке США по медицинским предметным рубрикам (MeSH)
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п Дада Р. (июнь 2019 г.). «Диагностика и лечение фолликулярной лимфомы: комплексный обзор» . Европейский журнал гематологии . 103 (3): 152–163. дои : 10.1111/ejh.13271 . ПМИД 31270855 .
- ^ Перейти обратно: а б с д и ж г час я дж к л Фишер Т., Зинг Н.П., Чиаттоне К.С., Федерико М., Луминари С. (январь 2018 г.). «Трансформированная фолликулярная лимфома». Анналы гематологии . 97 (1): 17–29. дои : 10.1007/s00277-017-3151-2 . hdl : 11380/1152780 . ПМИД 29043381 . S2CID 11524500 .
- ^ Перейти обратно: а б Ёсино Т., Таката К., Танака Т., Сато Ю., Тари А., Окада Х. (декабрь 2018 г.). «Недавний прогресс фолликулярной лимфомы в Японии и характеристики двенадцатиперстного типа». Международная патология . 68 (12): 665–676. дои : 10.1111/pin.12733 . ПМИД 30456840 . S2CID 53871784 .
- ^ Перейти обратно: а б с д и ж Линч Р.К., Гратцингер Д., Адвани Р.Х. (июль 2017 г.). «Клиническое влияние обновления классификации лимфом ВОЗ 2016 г.». Современные возможности лечения онкологии . 18 (7): 45. дои : 10.1007/s11864-017-0483-z . ПМИД 28670664 . S2CID 4415738 .
- ^ Перейти обратно: а б с д и ж г час я дж к л Буган К.М., Кайми П.Ф. (май 2019 г.). «Фолликулярная лимфома: диагностические и прогностические аспекты начального лечения». Текущие отчеты по онкологии . 21 (7): 63. дои : 10.1007/s11912-019-0808-0 . ПМИД 31119485 . S2CID 162181232 .
- ^ Перейти обратно: а б с д и ж г Оиси Н., Монтес-Морено С., Фельдман А.Л. (январь 2018 г.). «Неоплазия in situ при патологии лимфатических узлов». Семинары по диагностической патологии . 35 (1): 76–83. дои : 10.1053/j.semdp.2017.11.001 . ПМИД 29129357 .
- ^ Перейти обратно: а б Свердлов С.Х., Кампо Э., Пилери С.А., Харрис Н.Л., Стейн Х., Зиберт Р., Адвани Р., Гильмини М., Саллес Г.А., Зеленец А.Д., Яффе Э.С. (май 2016 г.). «Пересмотр классификации лимфоидных новообразований Всемирной организации здравоохранения 2016 г.» . Кровь . 127 (20): 2375–90. дои : 10.1182/blood-2016-01-643569 . ПМЦ 4874220 . ПМИД 26980727 .
- ^ Перейти обратно: а б с д и ж г час Таката К., Мията-Таката Т., Сато Ю., Ёсино Т. (2014). «Патология фолликулярной лимфомы» . Журнал клинической и экспериментальной гематопатологии . 54 (1): 3–9. дои : 10.3960/jslrt.54.3 . ПМИД 24942941 .
- ^ Перейти обратно: а б с д и ж г Ссылка БК (март 2018 г.). «Трансформация фолликулярной лимфомы – почему это происходит и можно ли это предотвратить?». Лучшие практики и исследования. Клиническая гематология . 31 (1): 49–56. дои : 10.1016/j.beha.2017.10.005 . ПМИД 29452666 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н Сориг М., Санчо Ж.М. (февраль 2018 г.). «Современные прогностические и прогностические факторы фолликулярной лимфомы». Анналы гематологии . 97 (2): 209–227. дои : 10.1007/s00277-017-3154-z . ПМИД 29032510 . S2CID 27602442 .
- ^ ЭнтрезГен 596
- ^ Карубе К., Скарфо Л., Кампо Э., Гиа П. (февраль 2014 г.). «Моноклональный В-клеточный лимфоцитоз и лимфома in situ». Семинары по биологии рака . 24 : 3–14. doi : 10.1016/j.semcancer.2013.08.003 . ПМИД 23999128 .
- ^ ЭнтрезГен 2146
- ^ ЭнтрезГен 1387
- ^ ЭнтрезГен 8740
- ^ ЭнтрезГен 8085
- ^ Карбоне А., Глогини А. (март 2014 г.). «Новые проблемы после распознавания фолликулярной лимфомы in situ». Лейкемия и лимфома . 55 (3): 482–90. дои : 10.3109/10428194.2013.807926 . ПМИД 23713483 . S2CID 39451928 .
- ^ ЭнтрезГен 7128
- ^ ЭнтрезГен 639
- ^ ЭнтрезГен 64121
- ^ Перейти обратно: а б с Гаскойн Р.Д., Надель Б., Паскуалуччи Л., Фитцгиббон Дж., Пэйтон Дж.Е., Мельник А. и др. (декабрь 2017 г.). «Фолликулярная лимфома: современный семинар ICML в Лугано, 2015 г.». Гематологическая онкология . 35 (4): 397–407. дои : 10.1002/hon.2411 . hdl : 11343/292747 . ПМИД 28378425 . S2CID 23980925 .
- ^ ЭнтрезГен 84433
- ^ EnterGene 974
- ^ EnterGene 965
- ^ EnterGene 1029
- ^ EnterGene 1030
- ^ ЭнтрезГен 8764
- ^ ЭнтрезГен 4609
- ^ Тобин Дж.В., Кин С., Гунавардана Дж., Молли П., Берч С., Хоанг Т., Ли Дж., Ли Л., Хуанг Л., Муринье В., Финк Дж.Л., Матигиан Н., Вари Ф., Фрэнсис С., Кридел Р., Вейгерт О., Хэбе С. , Юринович В., Клэппер В., Стейдл С., Сен Л.Х., Лоу С., Уайкс М.Н. и Ганди М.К. (декабрь 2019 г.). «Прогрессирование заболевания при фолликулярной лимфоме в течение 24 месяцев связано со снижением внутриопухолевой иммунной инфильтрации» . Дж. Клин Онкол . 37 (34): 3300–3309. дои : 10.1200/JCO.18.02365 . ПМК 6881104 . ПМИД 31461379 .
- ^ Перейти обратно: а б с д и ж г час Баргеци М, Бауманн Р, Коглиятти С, Дитрих ПЮ, Дючосал М, Гёде Дж, Хитц Ф, Конерманн С, Лори А, Мей У, Новак У, Папахристофилу А, Стеннер Ф, Таверна С, Зандер Т, Реннер С (2018) . «Диагностика и лечение фолликулярной лимфомы: обновленная информация» . Швейцарский медицинский еженедельник . 148 : w14635. дои : 10.4414/smw.2018.14635 . ПМИД 30044476 .
- ^ Перейти обратно: а б с д Таката К., Мията-Таката Т., Сато Ю., Ивамуро М., Окада Х., Тари А., Ёсино Т. (январь 2018 г.). «Желудочно-кишечная фолликулярная лимфома: современные знания и будущие проблемы». Международная патология . 68 (1): 1–6. дои : 10.1111/pin.12621 . ПМИД 29292593 . S2CID 206275496 .
- ^ Фукас П.Г., де Леваль Л. (январь 2015 г.). «Последние достижения в области кишечных лимфом». Гистопатология . 66 (1): 112–36. дои : 10.1111/его.12596 . ПМИД 25639480 . S2CID 20669863 .
- ^ Лайтнер А.Л., Шеннон Э., Гиббонс М.М., Рассел М.М. (апрель 2016 г.). «Первичная желудочно-кишечная неходжкинская лимфома тонкой и толстой кишки: систематический обзор». Журнал желудочно-кишечной хирургии . 20 (4): 827–39. дои : 10.1007/s11605-015-3052-4 . ПМИД 26676930 . S2CID 21756793 .
- ^ Перейти обратно: а б Пён С.И., Сон Г.А., Пэк Д.Х., Ким Г.Х., Ли Б.Е., Ли С.Дж., Юн Дж.Б., Хан С.И., Пак Д.Ю. (февраль 2017 г.). «Первичная фолликулярная лимфома прямой кишки, случайно обнаруженная при скрининговой колоноскопии» . Корейский журнал гастроэнтерологии = Тэхан Соваги Хакхое Чи . 69 (2): 139–142. дои : 10.4166/kjg.2017.69.2.139 . ПМИД 28239083 .
- ^ Маркс Э, Ши Ю (апрель 2018 г.). «Фолликулярная лимфома дуоденального типа: клинико-патологический обзор» . Архивы патологии и лабораторной медицины . 142 (4): 542–547. дои : 10.5858/arpa.2016-0519-RS . ПМИД 29565210 .
- ^ Вайндорф СК, Смит Л.Б., Оуэнс С.Р. (ноябрь 2018 г.). «Обновленная информация о желудочно-кишечных лимфомах» . Архивы патологии и лабораторной медицины . 142 (11): 1347–1351. дои : 10.5858/arpa.2018-0275-RA . ПМИД 30407861 .
- ^ Перейти обратно: а б Мой Б.Т., Уилмот Дж., Баллестерос Э., Форухар Ф., Вазири Х. (сентябрь 2016 г.). «Первичная фолликулярная лимфома желудочно-кишечного тракта: описание случая и обзор». Журнал рака желудочно-кишечного тракта . 47 (3): 255–63. дои : 10.1007/s12029-016-9847-z . ПМИД 27277664 . S2CID 1014977 .
- ^ Перейти обратно: а б с д и Вессманн В., Кинтанилья-Мартинес Л. (июнь 2019 г.). «Редкие зрелые В-клеточные лимфомы у детей и подростков» . Гематологическая онкология . 37 (Приложение 1): 53–61. дои : 10.1002/hon.2585 . ПМИД 31187530 .
- ^ Перейти обратно: а б с Ку М., Огами Р.С. (май 2017 г.). «Фолликулярная лимфома детского типа и детская узловая лимфома маргинальной зоны: последние клинические, морфологические, иммунофенотипические и генетические данные». Достижения анатомической патологии . 24 (3): 128–135. дои : 10.1097/PAP.0000000000000144 . ПМИД 28277421 .
- ^ Шукла В., Лу Р. (август 2014 г.). «IRF4 и IRF8: Управление достоинствами B-лимфоцитов» . Границы в биологии . 9 (4): 269–282. дои : 10.1007/s11515-014-1318-y . ПМК 4261187 . ПМИД 25506356 .
- ^ «Фактор регулирования интерферона 8 IRF8 [Homo sapiens (человек)] – Ген – NCBI» .
- ^ «MAP2K1 митоген-активируемая протеинкиназа-киназа 1 [Homo sapiens (человек)] – Ген – NCBI» .
- ^ Перейти обратно: а б Лоунс М.А., Рафаэль М., Маккарти К., Уотерспун А., Терьер-Лакомб М.Дж., Рамзи А.Д., Макленнан К., Каир М.С., Джеррард М., Мишон Дж., Пэтт С., Пинкертон Р., Сендер Л., Ауперин А., Спосто Р., Уэстон С., Херема Н.А., Сэнгер В.Г., фон Аллмен Д., Перкинс С.Л. (январь 2012 г.). «Первичная фолликулярная лимфома яичка у детей и подростков» . Журнал детской гематологии/онкологии . 34 (1): 68–71. дои : 10.1097/MPH.0b013e31820e4636 . ПМК 3251817 . ПМИД 22215099 .
- ^ Сюй Х, Яо Ф (март 2019 г.). «Первичная лимфома яичка: анализ SEER 1169 случаев» . Письма об онкологии . 17 (3): 3113–3124. дои : 10.3892/ol.2019.9953 . ПМК 6396186 . ПМИД 30867741 .
- ^ Чеа Сай, Вирт А., Сеймур Дж. Ф. (январь 2014 г.). «Первичная лимфома яичка» . Кровь . 123 (4): 486–93. дои : 10.1182/blood-2013-10-530659 . ПМИД 24282217 .
- ^ Лоссос, Исландия; Гаскойн, Р.Д. (2011). «Трансформация фолликулярной лимфомы» . Лучшие практики и исследования. Клиническая гематология . 24 (2): 147–63. дои : 10.1016/j.beha.2011.02.006 . ПМЦ 3112479 . ПМИД 21658615 .
- ^ Аладжио Р., Амадор К., Анагностопулос И., Аттигалле А.Д., Араужо ИБО, Берти Э.; и др. (2022). «5-е издание Классификации гематолимфоидных опухолей Всемирной организации здравоохранения: лимфоидные новообразования» . Лейкемия . 36 (7): 1720–1748. дои : 10.1038/s41375-022-01620-2 . ПМЦ 9214472 . ПМИД 35732829 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Вайсманн Д. «Фолликулярные лимфомы» . Университет медицины и стоматологии Нью-Джерси . Архивировано из оригинала 4 марта 2016 г. Проверено 26 июля 2008 г.
- ^ Перейти обратно: а б с Сориг М., Санчо Х.М. (май 2019 г.). «Недавние эпохальные исследования фолликулярной лимфомы». Обзоры крови . 35 : 68–80. дои : 10.1016/j.blre.2019.03.006 . ПМИД 30928169 . S2CID 89617933 .
- ^ Листер А. «Фолликулярная лимфома: перспектива, варианты лечения и стратегия» . МедСкейп .
- ^ Солаль-Селиньи П., Беллей М., Марчеселли Л., Пеше Э.А., Пилери С., Маклафлин П., Луминари С., Про Б, Монтото С., Феррери А.Дж., Деконинк Е., Милпьед Н., Гордон Л.И., Федерико М. (ноябрь 2012 г.). «Бдительное ожидание при фолликулярной лимфоме с низкой опухолевой нагрузкой в эпоху ритуксимаба: результаты базы данных исследования F2» . Журнал клинической онкологии . 30 (31): 3848–53. дои : 10.1200/JCO.2010.33.4474 . ПМИД 23008294 .
- ^ Ганапати К.А., Питталуга С., Одехиде О.О., Фридман А.С., Яффе Э.С. (сентябрь 2014 г.). «Ранние лимфоидные поражения: концептуальные, диагностические и клинические проблемы» . Гематологическая . 99 (9): 1421–32. дои : 10.3324/haematol.2014.107938 . ПМЦ 4562530 . ПМИД 25176983 .
- ^ Паванелло Ф., Стеффанони С., Гильмини М., Зукка Э. (2016). «Системная передовая терапия фолликулярной лимфомы: когда, кому и как» . Средиземноморский журнал гематологии и инфекционных заболеваний . 8 (1): e2016062. дои : 10.4084/MJHID.2016.062 . ПМК 5111519 . ПМИД 27872742 .
Внешние ссылки
[ редактировать ]- Запись о фолликулярной крупноклеточной лимфоме в общедоступном словаре терминов по раку NCI
| Базы данных органов управления : Национальные |
|---|