Миелодиспластический синдром
| Миелодиспластический синдром | |
|---|---|
| Другие имена | Прелейкемия, миелодисплазия [1] [2] |
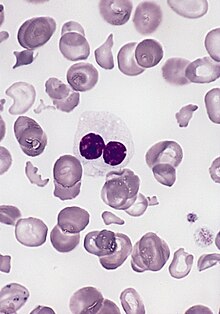 | |
| Мазок крови человека с миелодиспластическим синдромом. гипогранулярный нейтрофил с ядром псевдо-Пельгера-Хьюэ Показан . Встречаются также эритроциты аномальной формы , отчасти связанные с удалением селезенки . | |
| Специальность | Гематология , онкология |
| Симптомы | Нет, чувство усталости , одышка , легкое кровотечение , частые инфекции. [3] |
| Обычное начало | ~ 70 лет [4] |
| Факторы риска | Предыдущая химиотерапия , лучевая терапия , некоторые химические вещества, такие как табачный дым , пестициды и бензол , воздействие ртути или свинца. [3] |
| Метод диагностики | Анализ крови, биопсия костного мозга [3] |
| Уход | Поддерживающая терапия , лекарства, трансплантация стволовых клеток [3] |
| Медикамент | Леналидомид , антитимоцитарный глобулин , азацитидин [3] |
| Прогноз | Типичное время выживания 2,5 года. [3] |
Миелодиспластический синдром ( МДС ) относится к группе раковых заболеваний , при которых незрелые клетки крови в костном мозге не созревают и, как следствие, не развиваются в здоровые клетки крови. [3] На ранних стадиях обычно никаких симптомов не наблюдается. [3] Позже симптомы могут включать усталость , одышку , нарушение свертываемости крови , анемию или частые инфекции . [3] Некоторые типы могут перерасти в острый миелоидный лейкоз . [3]
Факторы риска включают предшествующую химиотерапию или лучевую терапию , воздействие определенных химических веществ, таких как табачный дым , пестициды и бензол , а также воздействие тяжелых металлов, таких как ртуть или свинец . [3] Проблемы с образованием клеток крови приводят к некоторому сочетанию низкого количества эритроцитов , тромбоцитов и лейкоцитов . [3] Некоторые типы МДС вызывают увеличение производства незрелых клеток крови (так называемых бластов ) в костном мозге или крови . [3] Различные типы МДС идентифицируются на основе особенностей изменений в клетках крови и костном мозге. [3]
Лечение может включать поддерживающую терапию , медикаментозную терапию и трансплантацию гемопоэтических стволовых клеток . [3] Поддерживающая терапия может включать переливание крови , лекарства, повышающие выработку эритроцитов , и антибиотики . [3] Лекарственная терапия может включать препараты леналидомид , антитимоцитарный глобулин и азацитидин . [3] Некоторых людей можно вылечить с помощью химиотерапии с последующей трансплантацией стволовых клеток от донора. [3]
Около семи из 100 000 человек страдают МДС; около четырех человек на 100 000 человек ежегодно заболевают этим заболеванием. [4] Типичный возраст начала заболевания — 70 лет. [4] Прогноз зависит от типа пораженных клеток, количества бластов в костном мозге или крови, а также изменений, присутствующих в хромосомах пораженных клеток. [3] Среднее время выживания после постановки диагноза составляет 2,5 года. [4] МДС был впервые признан в начале 1900-х годов; [5] в 1976 году его стали называть миелодиспластическим синдромом. [5]
Признаки и симптомы [ править ]

Признаки и симптомы неспецифичны и обычно связаны с цитопениями крови:
- Анемия (низкое количество эритроцитов или пониженный гемоглобин ) – хроническая усталость, одышка, ощущение озноба, иногда боль в груди. [6]
- Нейтропения (низкое количество нейтрофилов) – повышенная восприимчивость к инфекции. [7]
- Тромбоцитопения (низкое количество тромбоцитов) – повышенная склонность к кровотечениям и экхимозам (синякам), а также подкожным кровоизлияниям, приводящим к пурпуре или петехиям. [8] [9]
У многих людей симптомы отсутствуют, а цитопения крови или другие проблемы выявляются при обычном анализе крови: [10]
- Нейтропения, анемия и тромбоцитопения.
- Спленомегалия или редко гепатомегалия
- Аномальные гранулы в клетках, аномальная форма и размер ядра.
- Хромосомные аномалии , включая хромосомные транслокации и аномальное количество хромосом.
Хотя существует некоторый риск развития острого миелогенного лейкоза , около 50% смертей происходит в результате кровотечения или инфекции. Однако лейкемия, возникающая в результате миелодисплазии, как известно, устойчива к лечению. В раннем течении преобладает анемия. Большинство пациентов с симптомами жалуются на постепенное появление утомляемости и слабости, одышки и бледности , но по крайней мере у половины пациентов симптомы отсутствуют, и их МДС обнаруживается лишь случайно при рутинных анализах крови. человека Предыдущая химиотерапия или радиационное воздействие являются важным фактором в истории болезни . Лихорадка, потеря веса и спленомегалия должны указывать на миелодиспластическое/миелопролиферативное новообразование (МДС/МПН), а не на чистый миелодиспластический процесс. [11]
Причина [ править ]
Некоторые люди в анамнезе подвергались химиотерапии (особенно алкилирующим агентам, таким как мелфалан , циклофосфамид , бусульфан и хлорамбуцил ) или радиации (терапевтической или случайной) или тому и другому (например, во время трансплантации стволовых клеток по поводу другого заболевания). Работники некоторых отраслей промышленности, подвергающихся сильному воздействию углеводородов, например, нефтяной промышленности, имеют несколько более высокий риск заражения этим заболеванием, чем население в целом. Воздействие ксилола и бензола связано с миелодисплазией. Ветераны Вьетнама, подвергшиеся воздействию агента Orange, подвергаются риску развития МДС. Может существовать связь между развитием МДС «у выживших после атомной бомбардировки через 40–60 лет после радиационного воздействия» (в данном случае речь идет о людях, находившихся в непосредственной близости от места падения атомных бомб на Хиросиму и Нагасаки во время мировой войны). II). [12] Дети с синдромом Дауна восприимчивы к МДС, а семейный анамнез может указывать на наследственную форму сидеробластной анемии или анемии Фанкони . [13]
Патофизиология [ править ]
МДС чаще всего развивается без определенной причины. Факторы риска включают воздействие агентов, которые, как известно, вызывают повреждение ДНК, таких как радиация , бензол и некоторые виды химиотерапии; о других факторах риска сообщалось противоречиво. Доказать связь между предполагаемым заражением и развитием МДС может быть сложно, но наличие генетических аномалий может предоставить некоторую подтверждающую информацию. Вторичный МДС может возникать как поздняя токсичность противораковой терапии (МДС, связанный с терапией, т-МДС). МДС после воздействия радиации или алкилирующих агентов , таких как бусульфан, нитрозомочевина или прокарбазин , обычно возникает через 3–7 лет после воздействия и часто демонстрирует потерю хромосомы 5 или 7. МДС после воздействия ингибиторов ДНК-топоизомеразы II возникает после более короткого латентного периода, составляющего всего лишь 1–3 года и может иметь транслокацию 11q23. Другие ранее существовавшие заболевания костного мозга, такие как приобретенная апластическая анемия после иммуносупрессивного лечения и анемия Фанкони , могут перерасти в МДС. [13]
Считается, что МДС возникает в результате мутаций в мультипотентных стволовых клетках костного мозга , но конкретные дефекты, ответственные за эти заболевания, остаются плохо изученными. Нарушается дифференцировка значительное повышение уровня апоптотической клеток-предшественников крови, а в клетках костного мозга происходит гибели клеток. Клональная экспансия аномальных клеток приводит к образованию клеток, утративших способность к дифференцировке. Если общий процент миелобластов костного мозга превышает определенный порог (20% для ВОЗ и 30% для FAB трансформация в острый миелобластный лейкоз ), то говорят, что произошла (ОМЛ). Прогрессирование МДС в ОМЛ является хорошим примером многоступенчатой теории канцерогенеза, согласно которой серия мутаций происходит в изначально нормальной клетке и превращает ее в раковую клетку. [14]
Хотя признание лейкемической трансформации было исторически важным (см. «История» ), значительная часть заболеваемости и смертности, относимая на счет МДС, является результатом не трансформации в ОМЛ, а скорее цитопении, наблюдаемой у всех пациентов с МДС. Хотя анемия является наиболее распространенной цитопенией у пациентов с МДС, учитывая доступность переливания крови , пациенты с МДС редко страдают от тяжелой анемии. Двумя наиболее серьезными осложнениями у пациентов с МДС, возникающими в результате цитопении, являются кровотечение (из-за недостатка тромбоцитов) или инфекция (из-за отсутствия лейкоцитов). Длительное переливание эритроцитов приводит к перегрузке железом . [15]
Генетика [ править ]
Признание эпигенетических изменений в структуре ДНК при МДС объяснило успех двух (а именно гипометилирующих агентов 5-азацитидина и децитабина ) из трех (третий — леналидомид ) коммерчески доступных лекарств, одобренных Управлением по контролю за продуктами и лекарствами США для лечения МДС. Правильное метилирование ДНК имеет решающее значение для регуляции генов пролиферации, а потеря контроля метилирования ДНК может привести к неконтролируемому росту клеток и цитопениям. Недавно одобренные ингибиторы ДНК-метилтрансферазы используют этот механизм, создавая более упорядоченный профиль метилирования ДНК в гемопоэтических стволовых клеток ядре , тем самым восстанавливая нормальные показатели крови и замедляя прогрессирование МДС в острый лейкоз . [16]
Некоторые авторы предположили, что потеря митохондриальной функции с течением времени приводит к накоплению мутаций ДНК в гемопоэтических стволовых клетках, и это объясняет увеличение заболеваемости МДС у пожилых пациентов. Исследователи указывают на накопление митохондриальных отложений железа в кольцевом сидеробласте как на свидетельство митохондриальной дисфункции при МДС. [17]
Повреждение ДНК [ править ]
Считается, что старение гемопоэтических стволовых клеток связано с накоплением множественных генетических и эпигенетических аберраций, что позволяет предположить, что МДС частично связан с неспособностью адекватно справляться с повреждением ДНК . [18] Новая точка зрения заключается в том, что основным механизмом МДС может быть дефект одного или нескольких путей, которые участвуют в восстановлении поврежденной ДНК . [19] При МДС повышенная частота хромосомных разрывов указывает на дефекты процессов репарации ДНК. [20] Кроме того, повышенные уровни 8-оксогуанина были обнаружены в ДНК значительной части пациентов с МДС, что указывает на то, что путь эксцизионной репарации оснований , который участвует в обработке окислительных повреждений ДНК, может быть дефектным в этих случаях. [20]
5q-синдром [ править ]
, что делеция длинного плеча хромосомы 5 связана с диспластическими аномалиями гемопоэтических стволовых клеток. По крайней мере с 1974 г. известно [21] [22] К 2005 году леналидомид , химиотерапевтический препарат, был признан эффективным у пациентов с МДС с 5q-синдромом . [23] а в декабре 2005 года FDA США одобрило препарат по этому показанию. Пациенты с изолированным 5q-, низким риском IPSS и трансфузионной зависимостью лучше всего реагируют на леналидомид. Обычно прогноз для этих пациентов благоприятный, медиана выживаемости составляет 63 месяца. Леналидомид оказывает двойное действие: снижает количество злокачественных клонов у пациентов с 5q- и индуцирует лучшую дифференцировку здоровых эритроидных клеток, как это наблюдается у пациентов без делеции 5q. [ нужна ссылка ]
Мутации сплайсинга фактора
Мутации факторов сплайсинга обнаруживаются у 40–80% людей с МДС, при этом более высокая частота мутаций выявляется у людей, у которых больше кольцевых сидеробластов . [24]
IDH1 и IDH2 Мутации
Мутации в генах, кодирующих изоцитратдегидрогеназы 1 и 2 ( IDH1 и IDH2 ), встречаются у 10–20% пациентов с миелодиспластическим синдромом. [25] и ухудшают прогноз при МДС низкого риска. [26] Поскольку частота мутаций IDH1/2 увеличивается по мере увеличения злокачественности заболевания, эти результаты в совокупности позволяют предположить, что мутации IDH1/2 являются важными факторами прогрессирования МДС в более злокачественное состояние. [26]
Дефицит GATA2 [ править ]
Дефицит GATA2 — это группа заболеваний, вызванных дефектом, семейными или спорадическими инактивирующими мутациями в одном из двух GATA2 генов . Эти аутосомно-доминантные мутации вызывают снижение клеточного уровня продукта гена, GATA2. GATA2 Белок является транскрипционным фактором, имеющим решающее значение для эмбрионального развития , поддержания и функционирования кроветворных , лимфообразующих и других тканеобразующих стволовых клеток . В результате этих мутаций клеточные уровни GATA2 низкие, и у людей со временем развиваются гематологические, иммунологические, лимфатические или другие проявления. Среди этих проявлений выделяется МДС, который часто прогрессирует в острый миелоцитарный лейкоз или, реже, в хронический миеломоноцитарный лейкоз . [27] [28]
заболевание миелопролиферативное Транзиторное
Транзиторное миелопролиферативное заболевание представляет собой аномальную пролиферацию клона нераковых мегакариобластов в печени и костном мозге. Заболевание характерно только для лиц с синдромом Дауна или генетическими изменениями, сходными с таковыми при синдроме Дауна, развивается во время беременности или вскоре после рождения и разрешается в течение 3 месяцев или примерно в 10% случаев прогрессирует до острого мегакариобластного лейкоза . [29] [27] [30]
Диагностика [ править ]
Для диагностики миелодиспластического синдрома необходимо устранение других причин цитопении, а также дисплазии костного мозга, поэтому важно дифференцировать МДС от анемии, тромбоцитопении и лейкопении. [ нужна ссылка ]
Типичное диагностическое исследование включает в себя:
- Полный анализ крови и исследование мазка крови . Морфология мазка крови может дать представление о гемолитической анемии , скоплении тромбоцитов, приводящем к ложной тромбоцитопении , или лейкемии .
- Анализы крови для исключения других распространенных причин цитопении, таких как волчанка , гепатит , B12 дефицит , фолиевой кислоты или других витаминов , почечная или сердечная недостаточность , ВИЧ , гемолитическая анемия , моноклональная гаммапатия соответствующего возрасту возможность скрининга рака, : при всех анемиях следует рассмотреть . пациенты.
- Исследование костного мозга у гематопатолога : Это необходимо для установления диагноза, поскольку все гематопатологи считают диспластический костный мозг ключевым признаком миелодисплазии. [31]
- Цитогенетические или хромосомные исследования: в идеале это исследование проводится на аспирате костного мозга . Традиционная цитогенетика требует свежего образца, поскольку живые клетки заставляют вступать в метафазу, чтобы можно было увидеть хромосомы.
- Межфазное флуоресцентное гибридизационное тестирование in situ , обычно проводимое вместе с традиционным цитогенетическим тестированием , позволяет быстро обнаружить несколько хромосомных аномалий, связанных с МДС, включая del 5q, -7, +8 и del 20q.
- виртуальное кариотипирование . При МДС можно провести [32] который использует вычислительные инструменты для построения кариограммы из поврежденной ДНК. Виртуальное кариотипирование не требует культуры клеток и имеет значительно более высокое разрешение, чем традиционное цитогенетическое исследование, но не может обнаружить сбалансированные транслокации .
- Проточная цитометрия полезна для выявления бластов, аномального созревания миелоидных отложений и установления наличия любого лимфопролиферативного заболевания в костном мозге.
- Не следует упускать из виду тестирование на дефицит меди , поскольку он может морфологически напоминать МДС в биоптатах костного мозга. [33]
Признаками, обычно используемыми для определения МДС, являются цитопении крови, неэффективный гемопоэз, дизэритропоэз , дисгранулопоэз, дисмегакаропоэз и увеличение миелобластов.
Дисплазия может поражать все три линии, наблюдаемые в костном мозге. Лучший способ диагностировать дисплазию – по морфологии и специальным окраскам.( ПАСК ) используют для аспирата костного мозга и мазка периферической крови. Дисплазию миелоидного ряда определяют по:
- Гранулоцитарный ряд:
- Гиперсегментированные нейтрофилы (также наблюдаются при дефиците витамина B 12 /фолата)
- Гипосегментированные нейтрофилы ( псевдо Пельгера-Хюэ )
- Гипогранулярные нейтрофилы или псевдо Чедиак-Хигаси (крупные азурофильные гранулы )
- Палочки Ауэра – автоматически РАИБ II (при количестве бластов < 5% в периферической крови и < 10% в аспирате костного мозга); Также обратите внимание, что палочки Ауэра можно увидеть в зрелых нейтрофилах при ОМЛ с транслокацией t(8;21).
- Диморфные гранулы (базофильные и эозинофильные гранулы) внутри эозинофилов
- Эритроидная серия:
- Двуядерные предшественники эритроида и кариорексис
- Эритроидное ядерное почкование
- Эритроидные ядерные нити или межъядерные мостики (также наблюдаются при врожденных дизэритропоэтических анемиях )
- Потеря е-кадгерина в нормобластах является признаком отклонения.
- PAS (глобулярный в вакуолях или диффузное цитоплазматическое окрашивание) в предшественниках эритроида в аспирате костного мозга (не имеет отношения к биопсии костного мозга, фиксированной парафином). Примечание: PAS-вакуолярную позитивность можно увидеть в бластах L1 и L2 (классификация FAB; номенклатура L1 и L2 не используется в классификации ВОЗ).
- Кольцевые сидеробласты (10 или более гранул железа, окружающие одну треть или более ядра), видимые при окрашивании железа берлинским синим по Перлзу (> 15% кольцевых сидеробластов при подсчете среди предшественников эритроцитов при рефрактерной анемии с кольцевыми сидеробластами )
- Мегакариоцитарный ряд (может быть самый субъективный):
- Гипосегментированные ядерные особенности в мегакариоцитах, продуцирующих тромбоциты (отсутствие дольчатости)
- Гиперсегментированные ( остеокластические ) мегакариоциты.
- Раздувание тромбоцитов (видимо при интерференционно-контрастной микроскопии )
Другие пятна могут помочь в особых случаях (PAS и нафтол, ASD, положительная реакция хлорацетатэстеразы ) в эозинофилах является маркером аномалий, наблюдаемых при хроническом эозинофильном лейкозе , и признаком отклонения от нормы.
При биопсии костного мозга при дисплазии высокой степени (RAEB-I и RAEB-II) может наблюдаться атипичная локализация незрелых предшественников , которые представляют собой островки незрелых клеток-предшественников (миелобластов и промиелоцитов), локализованных в центре межтрабекулярного пространства, а не в центре межтрабекулярного пространства. прилегает к трабекулам или окружающим артериолам . Эту морфологию может быть трудно отличить от леченной лейкемии и восстановления незрелых нормальных элементов костного мозга. Кроме того, топографические изменения ядросодержащих эритроидных клеток можно наблюдать при ранней миелодисплазии ( РА и РАРС), когда нормобласты обнаруживаются рядом с костными трабекулами вместо формирования нормальных интерстициально расположенных эритроидных островков . [ нужна ссылка ]
Дифференциальный диагноз [ править ]
Миелодисплазия является диагнозом исключения и должна быть поставлена после правильного определения запасов железа, витаминов исключения дефицита врожденные заболевания, такие как врожденная дизэритропоэтическая анемия и питательных веществ. Кроме того, были выявлены (CDA I–IV), синдром Пирсона (сидеробластная анемия) , аномалия Жордана – вакуолизация во всех клеточных линиях, которая может наблюдаться при синдроме Чанарина-Дорфмана , дефицит фермента аминолевулиновой кислоты и другие. Известно, что эзотерические дефициты ферментов приводят к псевдомиелодиспластической картине в одной из клеточных линий; однако все три клеточные линии никогда не являются морфологически диспластическими у этих субъектов, за исключением хлорамфеникола, мышьяковой токсичности и других ядов. [ нужна ссылка ]
Все эти состояния характеризуются нарушениями производства одного или нескольких клеточных компонентов крови (эритроцитов, лейкоцитов, кроме лимфоцитов , и тромбоцитов или их клеток-предшественников, мегакариоцитов). [ нужна ссылка ]
Классификация [ править ]
Всемирная организация здравоохранения [ править ]
В конце 1990-х годов группа патологоанатомов и клиницистов, работающих под руководством Всемирной организации здравоохранения (ВОЗ), изменила эту классификацию, введя несколько новых категорий заболеваний и исключив другие. В 2008 году ВОЗ разработала новую схему классификации, которая в большей степени основана на генетических данных, но морфология клеток периферической крови, аспирата костного мозга и биопсии костного мозга по-прежнему являются скрининговыми тестами, используемыми для принятия решения о том, какая классификация лучше, а какая. цитогенетические аберрации могут быть связаны. [ нужна ссылка ]
В список диспластических синдромов по новой системе ВОЗ вошли:
| Миелодиспластический синдром | Описание |
|---|---|
| Рефрактерная цитопения с однолинейной дисплазией | Рефрактерная анемия, рефрактерная нейтропения и рефрактерная тромбоцитопения . |
| Рефрактерная анемия с кольцевыми сидеробластами (РАРС) | Тромбоцитоз (RARS-t) (предварительная форма), который по сути является миелодиспластическим/миелопролиферативным заболеванием и обычно имеет мутацию JAK2 (янус-киназа) – Новая классификация ВОЗ 2008 г. |
| Рефрактерная цитопения с многолинейной дисплазией (РКМД) | Включает подгруппу рефрактерной цитопении с многолинейной дисплазией и кольцевыми сидеробластами (RCMD-RS). RCMD включает пациентов с патологическими изменениями, не ограничивающимися эритроцитами (т.е. выраженной дисплазией предшественников лейкоцитов и предшественников тромбоцитов (мегакариоцитов). |
| Рефрактерная анемия с избытком бластов I и II. | РАИБ делился на бласты РАИБ-I (5–9% бластов) и РАИБ-II (10–19%), которые имеют худший прогноз, чем РАИБ-I. Палочки Ауэра можно увидеть при RAEB-II, который трудно отличить от острого миелолейкоза. |
| 5q- синдром | В классификацию был добавлен типичный синдром, наблюдаемый у пожилых женщин с нормальным или высоким количеством тромбоцитов и изолированными делециями длинного плеча хромосомы 5 в клетках костного мозга. |
| Миелодисплазия неклассифицируемая | Встречается в случаях дисплазии мегакариоцитов с фиброзом и других. |
| Рефрактерная цитопения детского возраста (дисплазия детского возраста) | – |
Примечание: не все врачи согласны с этой реклассификацией, поскольку основная патология этого заболевания недостаточно изучена.
синдром неклассифицированный Миелодиспластический
ВОЗ предложила критерии диагностики и классификации МДС, которые могут применяться в большинстве случаев. Однако отдельные случаи сложно отнести к определенным категориям из-за одной или нескольких необычных особенностей: [ нужна ссылка ]
- В редких случаях с уровнем взрыва менее 5% наблюдаются стержни Ауэра . Эти случаи обычно имеют черты RAMD .
- Иногда случаи МДС проявляются изолированной нейтропенией или тромбоцитопенией без анемии и диспластическими изменениями, ограниченными одной линией. Для описания этих случаев иногда использовались термины рефрактерная нейтропения и рефрактерная тромбоцитопения. Диагноз МДС у пациентов с нейтропенией или тромбоцитопенией без анемии следует ставить с осторожностью.
- наблюдается лейкоцитоз или тромбоцитоз . У пациентов с РА или РАИБ иногда вместо обычной цитопении
Управление [ править ]
Целями терапии являются контроль симптомов, улучшение качества жизни, улучшение общей выживаемости и замедление прогрессирования ОМЛ.
Рейтинг IPSS [34] Система может помочь сортировать пациентов для более агрессивного лечения (например, трансплантации костного мозга ), а также помочь определить наилучшие сроки этой терапии. [35] поддерживающая терапия препаратами крови и гемопоэтическими факторами роста (например, эритропоэтином Основой терапии является , нормативно-правовая база для использования эритропоэтинов развивается Medicare в США ). Согласно недавнему определению национального покрытия . Однако в этом документе не было сделано никаких комментариев по поводу использования гемопоэтических факторов роста при МДС. [36]
Агенты были одобрены Управлением по контролю за продуктами и лекарствами США (FDA) для лечения МДС:
- 5-азацитидин : медиана выживаемости в течение 21 месяца. [37] [38] [39] [40]
- Децитабин : уровень полного ответа достигает 43%. Исследование I фазы показало эффективность при ОМЛ при сочетании децитабина с вальпроевой кислотой . [41] [42] [43] [44]
- Леналидомид : эффективен для снижения потребности в переливании эритроцитов у пациентов с подтипом делеции хромосомы 5q при МДС. [45]
- Децитабин/цедазуридин с фиксированной дозировкой (Inqovi) представляет собой комбинированный препарат для лечения взрослых с миелодиспластическим синдромом (МДС) и хроническим миеломоноцитарным лейкозом (ХММЛ). [46]
Было показано, что химиотерапия гипометилирующими агентами 5-азацитидином и децитабином снижает потребность в переливании крови и замедляет прогрессирование МДС в ОМЛ. Леналидомид был одобрен FDA в декабре 2005 года только для применения при синдроме 5q . В США лечение МДС леналидомидом стоит около 9200 долларов в месяц. [47] Химиотерапию можно поддерживать другими препаратами, такими как полностью транс-ретиноевая кислота (ATRA), однако доказательства ее эффективности не ясны. [48]
Трансплантация HLA -совместимых аллогенных стволовых клеток , особенно у более молодых (т.е. менее 40 лет) и пациентов с более тяжелым поражением, открывает потенциал для радикальной терапии. Было обнаружено, что успех трансплантации костного мозга коррелирует с тяжестью МДС, определяемой по шкале IPSS, при этом пациенты с более благоприятной оценкой IPSS, как правило, имеют более благоприятный исход трансплантации. [49]
Уровни железа [ править ]
Перегрузка железом может развиться при МДС в результате переливания эритроцитов , которые являются основной частью поддерживающей терапии пациентов с анемией и МДС. Хотя специфическая терапия, которую получают пациенты, может в некоторых случаях облегчить потребность в переливании эритроцитов, многие пациенты с МДС могут не реагировать на это лечение, поэтому у них может развиться вторичный гемохроматоз из-за перегрузки железом в результате повторных переливаний эритроцитов.Пациенты, нуждающиеся в переливании относительно большого количества эритроцитов, могут испытывать неблагоприятное воздействие хронической перегрузки железом на функции печени, сердца и эндокринной системы.
У пациентов, нуждающихся в переливании большого количества эритроцитов ферритина в сыворотке , необходимо контролировать уровень , количество полученных переливаний эритроцитов и связанную с ними дисфункцию органов (сердце, печень и поджелудочная железа) для определения уровня железа. Мониторинг сывороточного ферритина также может быть полезен с целью снижения уровня ферритина до <1000 мкг/л два хелатора . В настоящее время в США доступны железа: дефероксамин для внутривенного применения и деферасирокс для перорального применения. Эти варианты теперь предоставляют потенциально полезные лекарства для лечения проблемы перегрузки железом. Третий хелатирующий агент доступен в Европе, деферипрон , для перорального применения, но недоступен в США. [ нужна ссылка ]
Клинические испытания препаратов, хелатирующих железо, на пациентах с МДС продолжаются, чтобы решить вопрос о том, изменяет ли хелирование железа естественную историю пациентов с МДС, которые зависят от переливания крови. устранение некоторых последствий перегрузки железом при МДС с помощью терапии хелаторами Было показано железа. И Фонд МДС, и Группа по составлению рекомендаций по МДС Национальной комплексной онкологической сети рекомендовали рассмотреть возможность применения хелаторной терапии для уменьшения перегрузки железом у отдельных пациентов с МДС. Данные также свидетельствуют о потенциальной ценности хелирования железа у пациентов, которым предстоит трансплантация стволовых клеток. Хотя деферасирокс, как правило, хорошо переносится (за исключением эпизодов желудочно-кишечных расстройств и дисфункции почек у некоторых пациентов), недавно к рекомендациям по лечению деферазироксом было добавлено предупреждение о безопасности FDA и Novartis. После постмаркетингового применения деферазирокса наблюдались редкие случаи острой почечной или печеночной недостаточности, некоторые из которых приводили к летальному исходу. В связи с этим пациенты, принимающие деферасирокс, должны находиться под тщательным наблюдением до начала терапии и регулярно в дальнейшем. [ нужна ссылка ]
Прогноз [ править ]
Прогноз при МДС варьируется: примерно у 30% пациентов развивается рефрактерный ОМЛ. Среднее время выживания варьируется от нескольких лет до месяцев, в зависимости от типа. Трансплантация стволовых клеток предлагает возможное излечение: выживаемость составляет 50% в течение 3 лет, хотя у пожилых пациентов дела обстоят хуже. [50]
Показатели хорошего прогноза :Младший возраст; нормальное или умеренно сниженное количество нейтрофилов или тромбоцитов; низкое количество бластов в костном мозге (<20%) и отсутствие бластов в крови; нет стержней Ауэра; кольцевые сидеробласты; нормальные или смешанные кариотипы без сложных хромосомных аномалий; и культура костного мозга in vitro с нелейкемическим характером роста
Признаки плохого прогноза :Преклонный возраст; тяжелая нейтропения или тромбоцитопения; высокое количество бластов в костном мозге (20–29%) или бласты в крови;стержни Ауэра; отсутствие кольцевых сидеробластов; аномальная локализация или незрелость предшественников гранулоцитов в срезе костного мозга;полностью или в основном аномальные кариотипы или сложные хромосомные аномалии костного мозга и культура костного мозга in vitro с лейкемическим характером роста
кариотипа Прогностические факторы :
- Хорошо: нормально, -Y, del(5q), del(20q)
- Промежуточный или переменный: +8, другие одиночные или двойные аномалии.
- Плохо: комплекс (>3 хромосомных аберраций); аномалии хромосомы 7 [51]
IPSS является наиболее часто используемым инструментом при МДС для прогнозирования долгосрочного результата. [52]
Цитогенетические аномалии могут быть обнаружены с помощью традиционной цитогенетики, панели FISH для МДС или виртуального кариотипа .
Наилучший прогноз наблюдается при РА и РАРС, при которых некоторые пациенты без трансплантации живут более десяти лет (обычно около трех-пяти лет, хотя возможна длительная ремиссия в случае успешной трансплантации костного мозга). Худший прогноз у РАЭБ-Т, где средняя продолжительность жизни составляет менее одного года. Примерно у четверти пациентов развивается манифестная лейкемия. Остальные умирают от осложнений, связанных с низким анализом крови или несвязанными заболеваниями. Международная система прогностической оценки — еще один инструмент для определения прогноза МДС, опубликованный в журнале Blood в 1997 году. [52] Эта система учитывает процент бластов в костном мозге, цитогенетику и количество цитопений.
маркеры Генетические
Хотя молекулярное профилирование геномов миелодиспластического синдрома еще формально не включено в общепринятые классификационные системы, оно улучшило понимание прогностических молекулярных факторов этого заболевания. Например, при МДС низкого риска мутации IDH1 и IDH2 связаны со значительным ухудшением выживаемости. [26]
Эпидемиология [ править ]
Точное количество людей с МДС неизвестно, поскольку он может остаться невыявленным и отслеживание синдрома не требуется. По некоторым оценкам, только в Соединенных Штатах ежегодно регистрируется от 10 000 до 20 000 новых случаев. Число новых случаев каждый год, вероятно, увеличивается по мере увеличения среднего возраста населения, и некоторые авторы предполагают, что число новых случаев среди людей старше 70 лет может достигать 15 на 100 000 в год. [53]
Типичный возраст постановки диагноза МДС составляет от 60 до 75 лет; некоторые люди моложе 50 лет, а у детей диагнозы редки. Мужчины болеют несколько чаще, чем женщины. [ нужна ссылка ]
История [ править ]
С начала 20-го века у некоторых людей с острым миелогенным лейкозом стали диагностировать предшествующий период анемии и аномальное производство клеток крови. Эти состояния были объединены с другими заболеваниями под термином «рефрактерная анемия». Первое описание «прелейкемии» как специфического заболевания было опубликовано в 1953 году Block et al. [54] Ранняя идентификация, характеристика и классификация этого расстройства были проблематичными, и у синдрома было много названий, пока в 1976 году не была опубликована классификация FAB, которая популяризировала термин МДС. [ нужна ссылка ]
Французско-американско-британская ( классификация ) FAB
В 1974 и 1975 годах группа патологоанатомов из Франции, США и Великобритании разработала первую широко используемую классификацию этих заболеваний. Эта французско-американско-британская классификация была опубликована в 1976 году. [55] и пересмотрен в 1982 году. Он использовался патологами и клиницистами почти 20 лет. Дела были разделены на пять категорий:
| МКБ-О | Имя | Описание |
|---|---|---|
| М9980/3 | Рефрактерная анемия (РА) | характеризуется наличием менее 5% примитивных клеток крови ( миелобластов ) в костном мозге и патологическими аномалиями, которые в первую очередь наблюдаются в предшественниках эритроцитов. |
| М9982/3 | Рефрактерная анемия с кольцевыми сидеробластами (РАРС) | также характеризуется наличием менее 5% миелобластов в костном мозге, но отличается наличием 15% или более предшественников эритроцитов в костном мозге, представляющих собой аномальные, наполненные железом клетки, называемые «кольцевыми сидеробластами». |
| М9983/3 | Рефрактерная анемия с избыточными бластами (РАИБ) | характеризуется 5–19% миелобластов в костном мозге |
| М9984/3 | Рефрактерная анемия с избытком бластов в трансформации (РАИБ-Т) | характеризуется наличием 5–19% миелобластов в костном мозге (более 20% бластов определяется как острый миелолейкоз ) |
| М9945/3 | Хронический миеломоноцитарный лейкоз (ХММЛ), не путать с хроническим миелогенным лейкозом или ХМЛ. | характеризуется менее 20% миелобластов в костном мозге и более 1*10 9 /L моноциты (разновидность лейкоцитов), циркулирующие в периферической крови. |
(Сравнительную таблицу можно получить в Кливлендской клинике . [56] )
Люди с МДС [ править ]
- Майкл Брекер , музыкант [57]
- Лаурентино Кортисо , президент Панамы [58]
- Роальд Даль , автор
- Нора Эфрон
- Джо Фаррелл , музыкант
- Пэт Хингл , актер [59]
- Джон Кирби , адвокат
- Джо Морган , бейсболист
- Пол Мотиан , музыкант [60]
- Джеймс В. Нэнс
- Робин Робертс , телеведущий
- Карл Саган , астрофизик [61]
- Сьюзен Зонтаг , автор [62]
- Фред Уиллард
- Нина Фош Актриса
См. также [ править ]
- Хлорома – тип опухоли, состоящей из лейкозных клеток.
- Клональный кроветворение
- Миелопролиферативный синдром
Ссылки [ править ]
- ^ «Миелодисплазия» . ПРОИДЧИК . Архивировано из оригинала 27 октября 2016 года . Проверено 27 октября 2016 г.
- ^ «Миелодиспластические синдромы» . НОРД (Национальная организация по редким заболеваниям) . Проверено 23 мая 2019 г.
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с «Лечение миелодиспластического синдрома (PDQ®) – версия для пациента» . НЦИ. 12 августа 2015 года. Архивировано из оригинала 5 октября 2016 года . Проверено 27 октября 2016 г.
- ↑ Перейти обратно: Перейти обратно: а б с д Герминг У., Коббе Г., Хаас Р., Гаттерманн Н. (ноябрь 2013 г.). «Миелодиспластические синдромы: диагностика, прогноз и лечение» . Немецкий международный медицинский журнал . 110 (46): 783–90. дои : 10.3238/arztebl.2013.0783 . ПМЦ 3855821 . ПМИД 24300826 .
- ↑ Перейти обратно: Перейти обратно: а б Хонг В.К., Холланд Дж.Ф. (2010). Медицина рака Холланд-Фрай (8-е изд.). PMPH-США. п. 1544. ИСБН 978-1-60795-014-1 . Архивировано из оригинала 27 октября 2016 г.
- ^ «Анемия: Обзор» . Библиотека медицинских концепций Lecturio . Проверено 15 августа 2021 г.
- ^ «Нейтропения» . Библиотека медицинских концепций Lecturio . Проверено 15 августа 2021 г.
- ^ Миелодиспластический синдром. Общество лейкемии и лимфомы. Уайт-Плейнс, штат Нью-Йорк. 2001. стр. 24. Дата обращения 12.05.2008.
- ^ «Тромбоцитопения» . Библиотека медицинских концепций Lecturio . Проверено 15 августа 2021 г.
- ^ «Миелодиспластические синдромы» . Библиотека медицинских концепций Lecturio . Проверено 11 августа 2021 г.
- ^ Патнаик М.М., Лашо Т. (04.12.2020). «Доказательный мини-обзор: синдромы перекрытия миелодиспластического синдрома и миелопролиферативных новообразований: целенаправленный обзор» . Гематология . 2020 (1): 460–4. doi : 10.1182/hematology.2020000163 . ПМЦ 7727594 . ПМИД 33275673 .
- ^ Иванага М., Сюй В.Л., Сода М., Такасаки Ю., Тавара М., Джо Т. и др. (февраль 2011 г.). «Риск развития миелодиспластических синдромов у людей, подвергшихся воздействию ионизирующей радиации: ретроспективное когортное исследование выживших после атомной бомбардировки Нагасаки». Журнал клинической онкологии . 29 (4): 428–34. дои : 10.1200/JCO.2010.31.3080 . ПМИД 21149671 .
- ↑ Перейти обратно: Перейти обратно: а б Докал И., Вуллиами Т. (август 2010 г.). «Наследственные синдромы недостаточности костного мозга» . Гематологическая . 95 (8): 1236–40. дои : 10.3324/haematol.2010.025619 . ПМЦ 2913069 . ПМИД 20675743 .
- ^ Бежар Р. (декабрь 2018 г.). «Какие биологические факторы предсказывают трансформацию в ОМЛ?» . Лучшие практики и исследования. Клиническая гематология . 31 (4): 341–5. дои : 10.1016/j.beha.2018.10.002 . ПМИД 30466744 . S2CID 53712886 .
- ^ Расел М., Махбуби С.К. (2022 г.), «Перегрузка трансфузионным железом» , StatPearls , Остров сокровищ (Флорида): StatPearls Publishing, PMID 32965817 , получено 3 февраля 2022 г.
- ^ Волах О, Стоун РМ (2015). «Как я лечу острый лейкоз смешанного фенотипа» . Кровь . 125 (16): 2477–85. дои : 10.1182/blood-2014-10-551465 . ПМИД 25605373 .
- ^ Каззола М., Инверницци Р., Бергамаски Г., Леви С., Корси Б., Траваглино Е. и др. (март 2003 г.). «Экспрессия митохондриального ферритина в эритроидных клетках пациентов с сидеробластной анемией» . Кровь . 101 (5): 1996–2000. дои : 10.1182/кровь-2002-07-2006 . ПМИД 12406866 . S2CID 5729203 .
- ^ Чжоу Т., Чен П., Гу Дж., Бишоп А.Дж., Скотт Л.М., Хэсти П. и др. (январь 2015 г.). «Потенциальная связь между неадекватным ответом на повреждение ДНК и развитием миелодиспластического синдрома» . Int J Mol Sci . 16 (1): 966–89. дои : 10.3390/ijms16010966 . ПМЦ 4307285 . ПМИД 25569081 .
- ^ Чжоу Т., Хэсти П., Уолтер К.А., Бишоп Эй.Дж., Скотт Л.М., Ребел VI (август 2013 г.). «Миелодиспластический синдром: неспособность адекватно реагировать на поврежденную ДНК?» . Эксп Гематол . 41 (8): 665–74. дои : 10.1016/j.exphem.2013.04.008 . ПМЦ 3729593 . ПМИД 23643835 .
- ↑ Перейти обратно: Перейти обратно: а б Янковская А.М., Гондек Л.П., Шпурка Х., Неарман З.П., Тиу Р.В., Мациевский Дж.П. (март 2008 г.). «Базовая дисфункция эксцизионного восстановления у подгруппы пациентов с миелодиспластическим синдромом». Лейкемия . 22 (3): 551–8. дои : 10.1038/sj.leu.2405055 . ПМИД 18059482 .
- ^ Банн HF (ноябрь 1986 г.). «5q- и нарушение кроветворения». Клиники гематологии . 15 (4): 1023–35. ПМИД 3552346 .
- ^ Ван ден Берге Х., Кассиман Дж. Дж., Дэвид Дж., Фринс Дж. П., Мишо Дж. Л., Сокал Дж. (октябрь 1974 г.). «Отчетливое гематологическое заболевание с делецией длинного плеча хромосомы № 5». Природа . 251 (5474): 437–38. Бибкод : 1974Natur.251..437V . дои : 10.1038/251437a0 . ПМИД 4421285 . S2CID 4286311 .
- ^ Список А, Куртин С., Роу Дж., Буреш А., Махадеван Д., Фукс Д. и др. (февраль 2005 г.). «Эффективность леналидомида при миелодиспластических синдромах» . Медицинский журнал Новой Англии . 352 (6): 549–57. doi : 10.1056/NEJMoa041668 . ПМИД 15703420 .
- ^ Розовский Ю, Китинг М, Эстров З (июль 2013 г.). «Значение мутаций сплайсосом при хроническом лимфоцитарном лейкозе» . Лейкозная лимфома . 54 (7): 1364–6. дои : 10.3109/10428194.2012.742528 . ПМК 4176818 . ПМИД 23270583 .
- ^ Моленаар Р.Дж., Радивоевич Т., Мациевски Дж.П., ван Норден К.Дж., Бликер Ф.Е. (декабрь 2014 г.). «Драйвер и пассажирские эффекты мутаций изоцитратдегидрогеназы 1 и 2 в онкогенезе и продлении выживаемости». Biochimica et Biophysical Acta (BBA) - Обзоры о раке . 1846 (2): 326–41. дои : 10.1016/j.bbcan.2014.05.004 . ПМИД 24880135 .
- ↑ Перейти обратно: Перейти обратно: а б с Моленаар Р.Дж., Тота С., Нагата Ю., Патель Б., Клементе М., Пшиходзен Б. и др. (ноябрь 2015 г.). «Клинические и биологические последствия наследственных и ненаследственных мутаций IDH1 и IDH2 в миелоидных новообразованиях» . Лейкемия . 29 (11): 2134–42. дои : 10.1038/leu.2015.91 . ПМК 5821256 . ПМИД 25836588 .
- ↑ Перейти обратно: Перейти обратно: а б Криспино Дж. Д., Хорвиц М. С. (апрель 2017 г.). «Мутации фактора GATA при гематологических заболеваниях» . Кровь . 129 (15): 2103–10. дои : 10.1182/blood-2016-09-687889 . ПМК 5391620 . ПМИД 28179280 .
- ^ Хирабаяши С., Влодарски М.В., Козыра Э., Нимейер К.М. (август 2017 г.). «Гетерогенность миелоидных новообразований, связанных с GATA2» . Международный журнал гематологии . 106 (2): 175–82. дои : 10.1007/s12185-017-2285-2 . ПМИД 28643018 .
- ^ Бхатнагар Н., Низери Л., Танстолл О., Вьяс П., Робертс И. (октябрь 2016 г.). «Транзиторные нарушения миелопоэза и ОМЛ при синдроме Дауна: обновленная информация» . Текущие отчеты о гематологических злокачественных новообразованиях . 11 (5): 333–41. дои : 10.1007/s11899-016-0338-x . ПМК 5031718 . ПМИД 27510823 .
- ^ Зивальд Л., Тауб Дж.В., Мэлони К.В., Маккейб Э.Р. (сентябрь 2012 г.). «Острые лейкозы у детей с синдромом Дауна». Молекулярная генетика и обмен веществ . 107 (1–2): 25–30. дои : 10.1016/j.ymgme.2012.07.011 . ПМИД 22867885 .
- ^ «Гематологическая клиника Рудхирам – Поиск в Google» . www.google.com . Проверено 3 февраля 2022 г.
- ^ Гондек Л.П., Тиу Р., О'Киф К.Л., Секерес М.А., Тейл К.С., Мациевски Дж.П. (февраль 2008 г.). «Хромосомные поражения и однородительская дисомия, обнаруженные с помощью массивов SNP при МДС, МДС/МПЛ и ОМЛ, вызванном МДС» . Кровь . 111 (3): 1534–42. дои : 10.1182/blood-2007-05-092304 . ПМК 2214746 . ПМИД 17954704 .
- ^ Хафф Дж.Д., Кеунг Ю.К., Такури М., Бити М.В., Херд Д.Д., Оуэн Дж. и др. (июль 2007 г.). «Дефицит меди вызывает обратимую миелодисплазию» . Американский журнал гематологии . 82 (7): 625–30. дои : 10.1002/ajh.20864 . ПМИД 17236184 . S2CID 44398996 .
- ^ «Marrowforums.org: МДС – миелодиспластические синдромы» . www.marrowforums.org . Архивировано из оригинала 14 июня 2011 года.
- ^ Катлер К.С., Ли С.Дж., Гринберг П., Диг Х.Дж., Перес В.С., Анасетти С. и др. (июль 2004 г.). «Анализ решений по аллогенной трансплантации костного мозга при миелодиспластических синдромах: отсроченная трансплантация при миелодисплазии низкого риска связана с улучшением результатов» . Кровь . 104 (2): 579–85. дои : 10.1182/кровь-2004-01-0338 . ПМИД 15039286 . S2CID 17907118 .
- ^ «Центры услуг Medicare и Medicaid» . Архивировано из оригинала 5 октября 2008 г. Проверено 29 октября 2007 г.
- ^ Вейерманс П., Любберт М., Верхуф Г., Босли А., Равут С., Андре М. и др. (март 2000 г.). «Низкие дозы 5-аза-2'-дезоксицитидина, гипометилирующего агента ДНК, для лечения миелодиспластического синдрома высокого риска: многоцентровое исследование II фазы у пожилых пациентов». Журнал клинической онкологии . 18 (5): 956–62. дои : 10.1200/JCO.2000.18.5.956 . ПМИД 10694544 .
- ^ Любберт М., Виерманс П., Кунцманн Р., Верхуф Г., Босли А., Равут С. и др. (август 2001 г.). «Цитогенетические реакции при миелодиспластическом синдроме высокого риска после лечения низкими дозами ингибитора метилирования ДНК 5-аза-2'-дезоксицитидина». Британский журнал гематологии . 114 (2): 349–57. дои : 10.1111/j.1365-2141.2001.02933.x . ПМИД 11529854 .
- ^ Сильверман Л.Р., Демакос Е.П., Петерсон Б.Л., Корнблит А.Б., Холланд Дж.К., Одчимар-Рейссиг Р. и др. (май 2002 г.). «Рандомизированное контролируемое исследование азацитидина у пациентов с миелодиспластическим синдромом: исследование группы B рака и лейкемии». Журнал клинической онкологии . 20 (10): 2429–40. дои : 10.1200/JCO.2002.04.117 . ПМИД 12011120 .
- ^ Сильверман Л.Р., Маккензи Д.Р., Петерсон Б.Л., Холланд Дж.Ф., Бэкстрем Дж.Т., Бич С.Л. и др. (август 2006 г.). «Дальнейший анализ исследований азацитидина у пациентов с миелодиспластическим синдромом: исследования 8421, 8921 и 9221 группы B по раку и лейкемии» . Журнал клинической онкологии . 24 (24): 3895–903. дои : 10.1200/JCO.2005.05.4346 . ПМИД 16921040 .
- ^ Кантарджян Х.М., О'Брайен С., Шан Дж., Ариби А., Гарсия-Манеро Г., Джаббур Э. и др. (январь 2007 г.). «Обновление опыта применения децитабина при миелодиспластическом синдроме повышенного риска и анализ прогностических факторов, связанных с исходом» . Рак . 109 (2): 265–73. дои : 10.1002/cncr.22376 . ПМИД 17133405 . S2CID 41205800 .
- ^ Кантарджян Х., Исса Дж.П., Розенфельд К.С., Беннетт Дж.М., Альбитар М., ДиПерсио Дж. и др. (апрель 2006 г.). «Децитабин улучшает результаты лечения пациентов с миелодиспластическими синдромами: результаты рандомизированного исследования III фазы» . Рак . 106 (8): 1794–803. дои : 10.1002/cncr.21792 . ПМИД 16532500 . S2CID 9556660 .
- ^ Кантарджян Х., Оки Ю., Гарсия-Манеро Дж., Хуанг Х., О'Брайен С., Кортес Дж. и др. (январь 2007 г.). «Результаты рандомизированного исследования 3 схем применения децитабина в низких дозах при миелодиспластическом синдроме высокого риска и хроническом миеломоноцитарном лейкозе» . Кровь . 109 (1): 52–57. дои : 10.1182/кровь-2006-05-021162 . ПМИД 16882708 .
- ^ Блюм В., Клисович Р.Б., Хакансон Б., Лю З., Лю С., Девайн Х. и др. (сентябрь 2007 г.). «Фаза I исследования децитабина отдельно или в сочетании с вальпроевой кислотой при остром миелолейкозе» . Журнал клинической онкологии . 25 (25): 3884–91. дои : 10.1200/JCO.2006.09.4169 . ПМИД 17679729 .
- ^ Список А, Девальд Г., Беннетт Дж., Гиагунидис А., Раза А., Фельдман Э. и др. (октябрь 2006 г.). «Леналидомид при миелодиспластическом синдроме с делецией хромосомы 5q» . Медицинский журнал Новой Англии . 355 (14): 1456–65. doi : 10.1056/NEJMoa061292 . ПМИД 17021321 .
- ^ «FDA одобрило новый метод лечения миелодиспластического синдрома (МДС), который можно принимать дома» . США Управление по контролю за продуктами и лекарствами (FDA) (пресс-релиз). 7 июля 2020 г. Проверено 7 июля 2020 г.
 В данную статью включен текст из этого источника, находящегося в свободном доступе .
В данную статью включен текст из этого источника, находящегося в свободном доступе . - ^ «Леналидомид (Ревлимид) при анемии миелодиспластического синдрома». Медицинское письмо о лекарствах и терапии . 48 (1232): 31–32. Апрель 2006 г. PMID 16625140 .
- ^ Кюлей-Багери И., Кройцер К.А., Монсеф И., Любберт М., Скец Н. и др. (Кокрейновская группа по гематологическим злокачественным новообразованиям) (август 2018 г.). «Эффекты полностью транс-ретиноевой кислоты (ATRA) в дополнение к химиотерапии у взрослых с острым миелоидным лейкозом (ОМЛ) (неострый промиелоцитарный лейкоз (не-APL))» . Кокрановская база данных систематических обзоров . 2018 (8): CD011960. дои : 10.1002/14651858.CD011960.pub2 . ПМК 6513628 . ПМИД 30080246 .
- ^ Остервельд М., Виттебол С.Х., Лемменс В.А., Кименей Б.А., Чатик А., Муус П. и др. (октябрь 2003 г.). «Влияние стратегии интенсивной противолейкемической терапии на прогноз пациентов с миелодиспластическим синдромом в возрасте до 61 года по группам риска Международной прогностической системы оценки» . Британский журнал гематологии . 123 (1): 81–89. дои : 10.1046/j.1365-2141.2003.04544.x . ПМИД 14510946 . S2CID 24037285 .
- ^ Каспер, Деннис Л., Браунвальд, Юджин, Фаучи, Энтони и др. (2005). Принципы внутренней медицины Харрисона (16-е изд.). Нью-Йорк: МакГроу-Хилл. п. 625 . ISBN 978-0-07-139140-5 .
- ^ Соле Ф., Эспинет Б., Санс Г.Ф., Сервера Дж., Каласанс М.Дж., Луньо Э. и др. (февраль 2000 г.). «Частота, характеристика и прогностическое значение хромосомных аномалий у 640 пациентов с первичными миелодиспластическими синдромами. Grupo Cooperativo Español de Citogenetica Hematologica». Британский журнал гематологии . 108 (2): 346–56. дои : 10.1046/j.1365-2141.2000.01868.x . ПМИД 10691865 . S2CID 10149222 .
- ↑ Перейти обратно: Перейти обратно: а б Гринберг П., Кокс С., ЛеБо М.М., Фено П., Морель П., Санс Г. и др. (март 1997 г.). «Международная балльная система оценки прогноза при миелодиспластических синдромах» . Кровь . 89 (6): 2079–88. дои : 10.1182/blood.V89.6.2079 . ПМИД 9058730 .
- ^ Аул С., Гиагунидис А., Герминг У. (июнь 2001 г.). «Эпидемиологические особенности миелодиспластических синдромов: результаты региональных онкологических исследований и больничная статистика». Международный журнал гематологии . 73 (4): 405–10. дои : 10.1007/BF02994001 . ПМИД 11503953 . S2CID 24340387 .
- ^ Блок М., Джейкобсон Л.О., Бетард В.Ф. (июль 1953 г.). «Предлейкемический острый лейкоз человека». Журнал Американской медицинской ассоциации . 152 (11): 1018–28. дои : 10.1001/jama.1953.03690110032010 . ПМИД 13052490 .
- ^ Беннетт Дж.М., Катовский Д., Дэниел М.Т., Фландрин Г., Гальтон Д.А., Гральник Х.Р. и др. (август 1976 г.). «Предложения по классификации острых лейкозов. Французско-американско-британская (FAB) кооперативная группа». Британский журнал гематологии . 33 (4): 451–58. дои : 10.1111/j.1365-2141.1976.tb03563.x . ПМИД 188440 . S2CID 9985915 .
- ^ «Таблица 1: Франко-американско-британская (FAB) классификация МДС» . Архивировано из оригинала 17 января 2006 г.
- ^ «Саксофонист Брекер умирает от МДС» . Разнообразие . 14 января 2007 года . Проверено 23 сентября 2018 г.
- ^ «Панама сообщает, что президент Кортисо все еще находится в стадии ремиссии от редкого заболевания крови» . Рейтер . 06.12.2023 . Проверено 6 декабря 2023 г.
- ^ Персонал J (4 января 2009 г.). «Актер-ветеран Пэт Хингл умер в возрасте 84 лет в своем доме в Северной Каролине» . Уинстон-Сейлемский журнал .
- ^ Макклеллан Д. (24 ноября 2011 г.). «Пол Мотиан умирает в 80 лет; джазовый барабанщик и композитор» . Лос-Анджелес Таймс . Архивировано из оригинала 14 апреля 2016 года . Проверено 22 февраля 2020 г. .
- ^ «Вспоминая Карла Сагана» . Вселенная сегодня . 9 ноября 2012 года. Архивировано из оригинала 12 марта 2017 года . Проверено 10 марта 2017 г.
- ^ «Болезнь как больше, чем метафора» . Журнал «Нью-Йорк Таймс» . 4 декабря 2005 г. Проверено 18 декабря 2017 г.
Внешние ссылки [ править ]
- Миелодиспластический синдром у Керли
- Фено П., Хаасе Д., Санс Г.Ф., Сантини В., Буске К. (сентябрь 2014 г.). «Миелодиспластические синдромы: Клинические рекомендации ESMO по диагностике, лечению и наблюдению». Энн Онкол . 25 (Приложение 3): iii57–69. дои : 10.1093/annonc/mdu180 . hdl : 2158/1078046 . ПМИД 25185242 .