Кариотип (рис . 1) — это характерный хромосомный набор эукариот видов . [3] [4] Кариотип обычно представляет собой изображение хромосом одной клетки, расположенных от самой большой (хромосома 1) к самой маленькой (хромосома 22), причем половые хромосомы (X и Y) показаны последними. Исторически кариотипы получали путем окрашивания клеток после того, как они были химически задержаны во время клеточного деления. Кариотипы использовались в течение нескольких десятилетий для выявления хромосомных аномалий как в зародышевых, так и в раковых клетках. Обычные кариотипы могут оценить весь геном на предмет изменений в структуре и количестве хромосом, но разрешение относительно грубое, с пределом обнаружения 5–10 МБ. [ нужна ссылка ]
Рис. 1. Кариотип человека мужского пола с использованием по Гимзе. окраски
платформы для создания кариотипов высокого разрешения in silico Недавно появились из разрушенной ДНК, такие как сравнительная геномная гибридизация массивов (arrayCGH) и массивы SNP . Концептуально массивы состоят из сотен и миллионов зондов, которые дополняют интересующую область генома. Разрушенная ДНК из тестируемого образца фрагментируется, метится и гибридизуется с массивом. Интенсивности сигналов гибридизации для каждого зонда используются специализированным программным обеспечением для генерации отношения log2 тестового/нормального для каждого зонда в массиве. [ нужна ссылка ]
Зная адрес каждого зонда в массиве и адрес каждого зонда в геноме, программное обеспечение выстраивает зонды в хромосомном порядке и реконструирует геном in silico (рис. 2 и 3).
Виртуальные кариотипы имеют значительно более высокое разрешение, чем традиционные цитогенетические методы. Фактическое разрешение будет зависеть от плотности датчиков на матрице. В настоящее время Affymetrix SNP6.0 представляет собой коммерчески доступный массив самой высокой плотности для приложений виртуального кариотипирования. Он содержит 1,8 миллиона полиморфных и неполиморфных маркеров с практическим разрешением 10–20 КБ — примерно размером гена. Это примерно в 1000 раз большее разрешение, чем у кариотипов, полученных с помощью традиционной цитогенетики. [ нужна ссылка ]
Виртуальные кариотипы могут быть выполнены на образцах зародышевой линии при конституциональных нарушениях. [5] [6] а клинические испытания доступны в десятках лабораторий, сертифицированных CLIA ( genetests.org ). Виртуальное кариотипирование также можно проводить на свежих или фиксированных формалином опухолях, залитых парафином. [7] [8] [9] Лаборатории, сертифицированные CLIA, предлагающие тестирование опухолей, включают Creighton Medical Laboratories (свежие образцы опухолей и образцы, залитые в парафин) и CombiMatrix Molecular Diagnostics (свежие образцы опухолей).
Рис. 2. Виртуальный кариотип образца хронического лимфоцитарного лейкоза с использованием массива SNP. Рис. 3. График лог2-отношения виртуального кариотипа образца хронического лимфоцитарного лейкоза с использованием массива SNP. Желтый = количество копий 2 (нормальный/диплоидный), голубой = 1 (делеция), розовый = 3 (трисомия).
Различные платформы для виртуального кариотипирования
Кариотипирование на основе массивов можно проводить с помощью нескольких различных платформ, как лабораторных, так и коммерческих. Сами массивы могут быть общегеномными (зонды распределены по всему геному) или таргетными (зонды для геномных областей, о которых известно, что они участвуют в конкретном заболевании) или комбинацией того и другого. Кроме того, в матрицах, используемых для кариотипирования, могут использоваться неполиморфные зонды, полиморфные зонды (т.е. содержащие SNP) или их комбинация. Неполиморфные зонды могут предоставить только информацию о количестве копий, тогда как массивы SNP могут предоставить как количество копий, так и статус потери гетерозиготности (LOH) в одном анализе. Типы зондов, используемые для неполиморфных массивов, включают кДНК, клоны BAC (например, BlueGnome ) и олигонуклеотиды (например, Agilent , Санта-Клара, Калифорния, США или Нимблеген , Мэдисон, Висконсин, США). Коммерчески доступные массивы SNP олигонуклеотидов могут быть твердофазными ( Affymetrix , Санта-Клара, Калифорния, США) или на основе шариков ( Illumina , Сан-Диего, Калифорния, США). Несмотря на разнообразие платформ, в конечном итоге все они используют геномную ДНК из разрушенных клеток для воссоздания кариотипа с высоким разрешением. в силиконе . Конечный продукт еще не имеет четкого названия и называется виртуальным кариотипированием. [8] [10] цифровое кариотипирование, [11] молекулярное аллелокариотипирование, [12] и молекулярное кариотипирование. [13] Другие термины, используемые для описания массивов, используемых для кариотипирования, включают SOMA (микрочипы SNP-олигонуклеотидов). [14] и CMA (хромосомный микрочип). [15] [16] Некоторые считают, что все платформы являются разновидностью сравнительной геномной гибридизации массивов (arrayCGH), в то время как другие резервируют этот термин для методов с двумя красителями, а третьи разделяют массивы SNP, поскольку они генерируют больше и другую информацию, чем методы arrayCGH с двумя красителями. [ нужна ссылка ]
Изменения количества копий можно увидеть как в образцах зародышевой линии, так и в образцах опухолей. Изменения количества копий могут быть обнаружены с помощью массивов с неполиморфными зондами, таких как arrayCGH, и массивов на основе SNP. Люди диплоидны, поэтому нормальное число копий неполовых хромосом всегда равно двум. [ нужна ссылка ]
Делеции: Делеция — это потеря генетического материала. Делеция может быть гетерозиготной (число копий 1) или гомозиготной (число копий 0, нуллисомия). Синдромы микроделеций являются примерами конституциональных нарушений, возникающих из-за небольших делеций в зародышевой ДНК. Делеции в опухолевых клетках могут представлять собой инактивацию гена-супрессора опухоли и иметь диагностическое, прогностическое или терапевтическое значение.
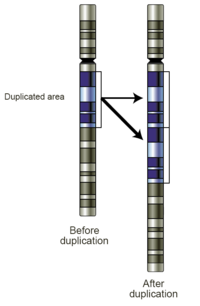
Прибыль: Увеличение количества копий представляет собой прирост генетического материала. Если получается всего лишь одна дополнительная копия сегмента ДНК, это можно назвать дупликацией (рис. 4). Если есть одна дополнительная копия всей хромосомы, это можно назвать трисомией . Увеличение числа копий в образцах зародышевой линии может быть связано с заболеванием или может быть доброкачественным вариантом числа копий . Когда они наблюдаются в опухолевых клетках, они могут иметь диагностическое, прогностическое или терапевтическое значение.
Рис. 4. Схема участка хромосомы до и после события дупликации.
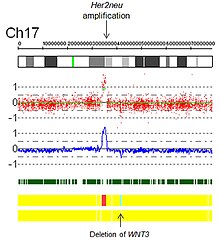
Амплификации. Технически амплификация — это тип увеличения количества копий, при котором число копий>10. В контексте биологии рака амплификации часто наблюдаются в онкогенах . Это может указывать на худший прогноз, помогать классифицировать опухоль или указывать на необходимость применения препарата. Примером пригодности препарата является амплификация Her2Neu и Герцептин , а также представлено изображение амплификации Her2Neu, обнаруженное с помощью виртуального кариотипирования массива SNP (рис. 5). Рис. 5. Амплификация Her2 с помощью виртуального кариотипа массива SNP.
Потеря гетерозиготности (LOH), автозиготных сегментов и однородительской дисомии.
Аутозиготные сегменты и однородительская дисомия (UPD) представляют собой диплоидные / «копически нейтральные» генетические данные и, следовательно, могут быть обнаружены только с помощью массивов на основе SNP. Как автозиготные сегменты, так и UPD будут демонстрировать потерю гетерозиготности (LOH) с числом копий, равным двум, согласно кариотипированию с использованием массива SNP. Термин «Профили гомозиготности» (ROH) является общим термином, который можно использовать как для автозиготных сегментов, так и для UPD. [ нужна ссылка ]
Аутозиготный сегмент: Аутозиготный сегмент является двуродительским и наблюдается только в зародышевой линии. Они представляют собой расширенные серии гомозиготных маркеров в геноме и возникают, когда идентичный блок гаплотипа унаследован от обоих родителей. Их также называют сегментами « идентичными по происхождению » (IBD), и их можно использовать для картирования гомозиготности. [17] [18]
Однородительская дисомия: UPD возникает, когда обе копии гена или геномной области унаследованы от одного и того же родителя. Это однородительский сегмент, в отличие от автозиготных сегментов, которые являются двуродительскими. Когда они присутствуют в зародышевой линии, они могут быть безвредными или ассоциироваться с заболеваниями, такими как Прадера-Вилли или синдромы Ангельмана . Также в отличие от аутозиготности UPD может развиваться в опухолевых клетках, и в литературе это называется приобретенным UPD или копийно-нейтральным LOH (рис. 6). Рис. 6. Копирование нейтральной LOH/однородительской дисомии. Приобретенная UPD довольно часто встречается как в гематологических, так и в солидных опухолях и, как сообщается, составляет от 20 до 80% LOH, наблюдаемых в опухолях человека. [19] [20] [21] [22] Приобретенный UPD может служить вторым хитом в гипотезе опухолевого генеза Кнудсона с двумя ударами и, таким образом, может быть биологическим эквивалентом делеции. [23] Поскольку этот тип поражения не может быть обнаружен с помощью arrayCGH, FISH или традиционной цитогенетики, для виртуального кариотипирования опухолей предпочтительны массивы на основе SNP.
Рис. 7. Виртуальный кариотип колоректальной карциномы (просмотр всего генома), демонстрирующий делеции, приросты, амплификации и приобретенные UPD (копийно-нейтральные LOH).
На рисунке 7 представлен виртуальный кариотип массива SNP колоректальной карциномы, демонстрирующий делеции, приросты, амплификации и приобретенный UPD (копировально-нейтральный LOH).
Виртуальный кариотип может быть создан практически из любой опухоли, но клиническое значение выявленных геномных аберраций различно для каждого типа опухоли. Клиническая полезность варьируется, и целесообразность лучше всего определяет онколог или патологоанатом после консультации с заведующим лабораторией, выполняющей виртуальный кариотип. Ниже приведены примеры типов рака, для которых клинические последствия конкретных геномных аберраций хорошо известны. Этот список является репрезентативным, а не исчерпывающим. На веб-сайте лаборатории цитогенетики Лаборатории гигиены штата Висконсин приведены дополнительные примеры клинически значимых генетических изменений, которые легко обнаружить с помощью виртуального кариотипирования. [1]
На основе серии из 493 образцов нейробластомы было сообщено, что общий геномный паттерн, проверенный с помощью кариотипирования на основе массивов, является предиктором исхода нейробластомы: [24]
Опухоли, демонстрирующие исключительно изменения числа копий всей хромосомы, были связаны с отличной выживаемостью.
Опухоли с любыми изменениями числа копий сегментарных хромосом были связаны с высоким риском рецидива.
В опухолях, демонстрирующих сегментарные изменения, дополнительными независимыми предикторами снижения общей выживаемости были амплификация MYCN, делеции 1p и 11q и прирост 1q.
В более ранних публикациях нейробластомы были разделены на три основных подтипа на основе цитогенетического профиля: [25]
Подтип 1: благоприятная нейробластома с почти триплоидией и преобладанием численных приростов и потерь, в основном представляющая неметастатические стадии 1, 2 и 4S НБ.
Подтипы 2A и 2B: встречаются при неблагоприятной распространенной нейробластоме, стадии 3 и 4, с потерей 11q и усилением 17q без амплификации MYCN (подтип 2A) или с амплификацией MYCN, часто вместе с делецией 1p и усилением 17q (подтип 2B).
Опухоспецифическая потеря гетерозиготности (LOH) для хромосом 1p и 16q идентифицирует подгруппу пациентов с опухолью Вильмса , у которых значительно повышен риск рецидива и смерти. LOH для этих хромосомных областей теперь можно использовать в качестве независимого прогностического фактора вместе со стадией заболевания, чтобы нацелить интенсивность лечения на риск неудачи лечения. [26] [27]
Кариотипирование на основе массивов можно использовать для выявления характерных хромосомных аберраций в опухолях почек со сложной морфологией. [8] [10] Кариотипирование на основе массивов хорошо работает при опухолях, залитых парафином. [29] и пригоден для рутинного клинического использования.
Кроме того, недавняя литература указывает на то, что определенные хромосомные аберрации связаны с исходом определенных подтипов эпителиальных опухолей почки. [30] Светлоклеточный рак почки: del 9p и del 14q являются плохими прогностическими показателями. [31] [32] Папиллярный почечно-клеточный рак: дупликация 1q указывает на фатальное прогрессирование. [33]
Кариотипирование на основе массивов является экономически эффективной альтернативой FISH для выявления хромосомных аномалий при хроническом лимфоцитарном лейкозе (ХЛЛ). Несколько клинических проверочных исследований показали соответствие >95% стандартной панели FISH для ХЛЛ. [12] [34] [35] [36] [37] Кроме того, многие исследования с использованием кариотипирования на основе массивов выявили «атипичные делеции», пропущенные стандартными зондами FISH, и приобретенную однородительскую дисомию в ключевых локусах для прогностического риска при ХЛЛ. [38] [39]
В клетках ХЛЛ выявлены четыре основных генетических отклонения, которые оказывают существенное влияние на поведение при заболевании. [40]
Делеции части короткого плеча хромосомы 17 (del 17p), нацеленной на р53, особенно вредны. Пациенты с этой аномалией имеют значительно более короткий интервал, прежде чем им потребуется терапия, и более короткую выживаемость. Эта аномалия обнаруживается у 5–10% пациентов с ХЛЛ.
Делеции длинного плеча хромосомы 11 (del 11q) также неблагоприятны, хотя и не в такой степени, как при del 17p. Аномалия нацелена на ген ATM и встречается нечасто при ХЛЛ (5–10%).
Трисомия 12, дополнительная хромосома 12, является относительно частой находкой, встречающейся у 20–25% пациентов и определяющей промежуточный прогноз.
Делеция 13q14 (del 13q14) является наиболее распространенной аномалией при ХЛЛ: примерно у 50% пациентов клетки содержат этот дефект. Когда del 13q14 наблюдается изолированно, пациенты имеют лучший прогноз, и большинство из них проживут многие годы, даже десятилетия, без необходимости лечения.
Авет-Луазо и др. в журнале Journal of Clinical Oncology использовали кариотипирование с использованием массива SNP 192 образцов множественной миеломы (ММ) для выявления генетических поражений, связанных с прогнозом, которые затем были подтверждены в отдельной когорте (n = 273). [41] При ММ отсутствие пролиферативного клона делает традиционную цитогенетику информативной лишь в ~30% случаев. Панели FISH полезны при ММ, но стандартные панели не позволяют обнаружить несколько ключевых генетических аномалий, о которых сообщалось в этом исследовании. [ нужна ссылка ]
Виртуальное кариотипирование выявило хромосомные аномалии в 98% случаев ММ.
del(12p13.31) является независимым неблагоприятным маркером.
amp(5q31.1) является благоприятным маркером
Прогностическое влияние amp(5q31.1) превосходит влияние гипердиплоидии, а также выявляет пациентов, которым терапия высокими дозами приносит большую пользу.
Кариотипирование на основе массивов не может обнаружить сбалансированные транслокации, такие как t(4;14), наблюдаемые примерно в 15% случаев ММ. Следовательно, FISH для этой транслокации также следует выполнять при использовании массивов SNP для обнаружения изменений количества копий по всему геному, имеющих прогностическое значение при ММ. [ нужна ссылка ]
на основе массивов, Кариотипирование 260 медуллобластом проведенное Pfister S и соавт. в результате были выделены следующие клинические подгруппы на основе цитогенетических профилей: [42]
Прогноз плохой: увеличение 6q или усиление MYC или MYCN.
Промежуточный уровень: усиление 17q или i(17q) без усиления 6q или усиление MYC или MYCN.
Совместная делеция 1p/19q считается «генетической подписью» олигодендроглиомы . Утрата аллелей 1p и 19q, по отдельности или в сочетании, чаще встречается в классических олигодендроглиомах, чем в астроцитомах или олигоастроцитомах. [43] В одном исследовании классические олигодендроглиомы показали потерю 1p в 35 из 42 (83%) случаев, потерю 19q в 28 из 39 (72%), и они сочетались в 27 из 39 (69%) случаев; не было значительной разницы в потере статуса гетерозиготности 1p/19q между олигодендроглиомами низкой степени злокачественности и анапластическими олигодендроглиомами. [43] Совместная делеция 1p/19q коррелирует как с химиочувствительностью, так и с улучшением прогноза при олигодендроглиомах. [44] [45] Большинство крупных онкологических центров регулярно проверяют наличие делеции 1p/19q в отчете о патологии олигодендроглиомы. Статус локусов 1p/19q можно определить с помощью FISH или виртуального кариотипирования. Преимущество виртуального кариотипирования заключается в том, что за один анализ можно оценить весь геном, а также локусы 1p/19q. Это позволяет оценить другие ключевые локусы в глиальных опухолях, такие как статус количества копий EGFR и TP53. [ нужна ссылка ]
В то время как прогностическая значимость делеций 1p и 19q хорошо известна для анапластических олигодендроглиом и смешанных олигоастроцитом, прогностическая значимость делеций для глиом низкой степени злокачественности более противоречива. Что касается глиом низкой степени злокачественности, недавнее исследование также предполагает, что совместная делеция 1p/19q может быть связана с транслокацией (1;19)(q10;p10), которая, как и комбинированная делеция 1p/19q, связана с превосходным общая выживаемость и выживаемость без прогрессирования у пациентов с глиомой низкой степени злокачественности. [46] В олигодендроглиомах лишь в редких случаях обнаруживаются мутации гена р53, в отличие от других глиом. [47] Амплификация рецептора эпидермального фактора роста и полная коделяция 1p/19q являются взаимоисключающими и предсказывают совершенно разные исходы, при этом амплификация EGFR предсказывает плохой прогноз. [48]
Инь и др. [49] изучили 55 глиобластомы клеточных линий и 6 линий GBM с использованием кариотипирования с использованием массива SNP. Приобретенный УПД был идентифицирован на уровне 17р в 13 из 61 случаев. Значительно сокращенное время выживания было обнаружено у пациентов с делецией 13q14 (RB) или делецией 17p13.1 (p53)/приобретенным UPD. В совокупности эти результаты позволяют предположить, что этот метод является быстрым, надежным и недорогим методом выявления общегеномных аномалий при ГБМ. Поскольку кариотипирование с использованием массива SNP можно выполнять на опухолях, залитых парафином, это является привлекательным вариантом, когда опухолевые клетки не растут в культуре для метафазной цитогенетики или когда желание кариотипирования возникает после фиксации образца формалином. [ нужна ссылка ]
Среди пациентов с аномалией 17p ~50% были делециями и ~50% были аУПД.
И 17p del, и 17p UPD были связаны с худшим исходом.
У 9 из 13 были гомозиготные мутации TP53, лежащие в основе UPD 17p.
Кроме того, в случаях неопределенной степени морфологии геномное профилирование может помочь в диагностике.
Сопутствующий прирост 7 баллов и потеря 10 баллов по существу патогномоничны для ГБМ. [50]
Амплификация EGFR, потеря PTEN (на 10q) и потеря p16 (на 9p) встречаются почти исключительно при глиобластоме и могут помочь отличить анапластическую астроцитому от глиобластомы. [51]
Цитогенетика , изучение характерных больших изменений в хромосомах раковых клеток , все чаще признается важным предиктором исхода острого лимфобластного лейкоза (ОЛЛ). [52] NB: Сбалансированные транслокации не могут быть обнаружены с помощью кариотипирования на основе массива (см. Ограничения ниже).
Некоторые цитогенетические подтипы имеют худший прогноз, чем другие. К ним относятся:
Транслокация между хромосомами 4 и 11 встречается примерно в 4% случаев и чаще всего встречается у детей младше 12 месяцев.
Не все транслокации хромосом имеют худший прогноз. Некоторые транслокации относительно благоприятны. Например, гипердиплоидия (>50 хромосом) является хорошим прогностическим фактором.
Полногеномную оценку изменений количества копий можно выполнить с помощью традиционной цитогенетики или виртуального кариотипирования. Виртуальное кариотипирование массива SNP может обнаруживать изменения количества копий и статус LOH, тогда как arrayCGH может обнаруживать только изменения количества копий. Копировально-нейтральная LOH (приобретенная однородительская дисомия) была обнаружена в ключевых локусах при ОЛЛ, таких как ген CDKN2A в положении 9p, которые имеют прогностическое значение. [53] [54] [55] Виртуальное кариотипирование с использованием массива SNP позволяет легко обнаружить нейтральный к копированию LOH. Массив CGH, FISH и традиционные цитогенетические методы не могут обнаружить копировально-нейтральный LOH.
Миелодиспластический синдром (МДС) обладает значительной клинической, морфологической и генетической гетерогенностью. Цитогенетика играет решающую роль в основанной на классификации Международной прогностической системе оценки (IPSS) МДС Всемирной организации здравоохранения. [57] [58]
Плохой прогноз: сложные аномалии (т.е. >=3 аномалий), −7 или del(7q).
Промежуточный прогноз: все другие аномалии, включая трисомию 8 и del(11q).
При сравнении метафазной цитогенетики, панели FISH и кариотипирования с использованием массива SNP при МДС было обнаружено, что каждый метод обеспечивает одинаковую диагностическую эффективность. Ни один метод не выявил всех дефектов, а уровень обнаружения увеличился примерно на 5% при использовании всех трех методов. [59]
Приобретенный UPD, который не выявляется с помощью FISH или цитогенетики, был зарегистрирован в нескольких ключевых локусах при МДС с использованием кариотипирования с использованием массива SNP, включая делецию 7/7q. [60] [61]
Негативные по филадельфийской хромосоме миелопролиферативные новообразования (МПН), включая истинную полицитемию, эссенциальную тромбоцитемию и первичный миелофиброз, демонстрируют присущую им тенденцию к трансформации в лейкемию (МПН-бластная фаза), которая сопровождается приобретением дополнительных геномных поражений.При исследовании 159 случаев [62] Анализ SNP-массива позволил выявить практически все цитогенетические аномалии и выявить дополнительные поражения с потенциально важными клиническими последствиями. [ нужна ссылка ]
Число геномных изменений в бластной фазе было более чем в 2–3 раза больше, чем в хронической фазе заболевания.
Делеция 17p (TP53) была в значительной степени связана с предшествующим воздействием гидроксимочевины, а также со сложным кариотипом в образцах с MPN-бластным кризисом. И делеция, и копия-нейтральный LOH 17p были связаны со сложным кариотипом, плохим прогностическим маркером при миелоидных злокачественных новообразованиях. Копировально-нейтральный LOH (приобретенный UPD) легко выявляется с помощью кариотипа массива SNP, но не с помощью цитогенетики, FISH или массива CGH.
Пациенты в бластной фазе с потерей хромосомного материала на 7q показали плохую выживаемость. Известно, что потеря 7q является прогностическим фактором быстрого прогрессирования и плохого ответа на терапию ОМЛ. Пациенты с фазой MPN-бласта с цитогенетически неопределяемой копией 7q нейтрального LOH имели сопоставимые показатели выживаемости с пациентами с 7/7q в лейкозных клетках.
Нейтральная копия 9p с гомозиготной мутацией JAK2 также была связана с худшим исходом при MPN-бластном кризе по сравнению с пациентами с гетерозиготным JAK2V617F или JAK2 дикого типа. В отличие от LOH на 17p, прогностическое влияние 9pCNN-LOH не зависело от установленных факторов риска, таких как 7/7q, 5q или сложный кариотип.
Идентификация биомаркеров колоректального рака особенно важна для пациентов со II стадией заболевания, у которых менее 20% случаев имеют рецидив опухоли. 18q LOH является признанным биомаркером, связанным с высоким риском рецидива опухоли при раке толстой кишки II стадии. [63] На рисунке 7 показан кариотип колоректальной карциномы с массивом SNP (полный геном).
Колоректальный рак классифицируют на специфические фенотипы опухолей на основе молекулярных профилей. [63] которые могут быть интегрированы с результатами других вспомогательных тестов, таких как тестирование нестабильности микросателлитов, IHC и статус мутации KRAS:
Хромосомная нестабильность (CIN), которая имеет аллельный дисбаланс в ряде хромосомных локусов, включая 5q, 8p, 17p и 18q (рис. 7).
Микросателлитная нестабильность (MSI), которая имеет тенденцию иметь диплоидный кариотип.
Злокачественные рабдоидные опухоли — редкие, высокоагрессивные новообразования, чаще всего встречающиеся у младенцев и детей раннего возраста. Из-за их гетерогенных гистологических особенностей диагностика часто может быть затруднена и может произойти неправильная классификация. В этих опухолях ген INI1 (SMARCB1) на хромосоме 22q действует как классический ген-супрессор опухоли. Инактивация INI1 может происходить посредством делеции, мутации или приобретенного UPD. [64]
В недавнем исследовании [64] Кариотипирование с помощью массива SNP выявило делеции или LOH 22q в 49/51 рабдоидных опухолях. Из них 14 были копийно-нейтральными LOH (или приобретенными UPD), что можно обнаружить с помощью кариотипирования с использованием массива SNP, но не с помощью FISH, цитогенетики или arrayCGH. MLPA обнаружила гомозиготную делецию одного экзона в одном образце, разрешение которой было ниже разрешения массива SNP. [ нужна ссылка ]
Кариотипирование с использованием массива SNP можно использовать, чтобы отличить, например, медуллобластому с изохромосомой 17q от первичной рабдоидной опухоли с утратой 22q11.2. При наличии показаний можно затем использовать молекулярный анализ INI1 с использованием MLPA и прямое секвенирование. После обнаружения связанных с опухолью изменений можно провести анализ зародышевой ДНК пациента и родителей, чтобы исключить наследственную или de novo мутацию зародышевой линии или делецию INI1, чтобы можно было провести соответствующую оценку риска рецидива. [64]
Наиболее важным генетическим изменением, связанным с плохим прогнозом при увеальной меланоме, является потеря полной копии хромосомы 3 ( моносомия 3), которая тесно коррелирует с распространением метастазов. [65] Прирост хромосом 6 и 8 часто используется для уточнения прогностической ценности скрининга моносомии 3: прирост 6p указывает на лучший прогноз, а прирост 8q указывает на худший прогноз при опухолях с дисомией 3. [66] В редких случаях опухоли с моносомией 3 могут дублировать оставшуюся копию хромосомы, возвращаясь в дисомное состояние, называемое изодисомией . [67] Изодосомия 3 прогностически эквивалентна моносомии 3, и обе могут быть обнаружены с помощью тестов на потерю гетерозиготности хромосомы 3 . [68]
В отличие от кариотипов, полученных с помощью традиционной цитогенетики, виртуальные кариотипы реконструируются с помощью компьютерных программ с использованием сигналов, полученных из разрушенной ДНК. По сути, компьютерная программа будет корректировать транслокации, выстраивая сигналы в хромосомном порядке. Следовательно, виртуальные кариотипы не могут обнаружить сбалансированные транслокации и инверсии . Они также могут обнаруживать генетические аберрации только в тех участках генома, которые представлены зондами на массиве. Кроме того, виртуальные кариотипы генерируют относительное число копий, нормализованное по отношению к диплоидному геному, поэтому тетраплоидные геномы будут конденсироваться в диплоидное пространство, если не будет выполнена перенормировка. Для перенормировки требуется вспомогательный клеточный анализ, например FISH, если используется arrayCGH. Для кариотипов, полученных из массивов на основе SNP, о тетраплоидии часто можно судить по сохранению гетерозиготности в области очевидной потери числа копий. [22] Мозаицизм низкого уровня или небольшие субклоны могут быть не обнаружены виртуальными кариотипами, поскольку присутствие нормальных клеток в образце ослабляет сигнал от аномального клона. Точная точка отказа с точки зрения минимального процента неопластических клеток будет зависеть от конкретной платформы и используемых алгоритмов. Многие программы для анализа числа копий, используемые для создания кариотипов на основе массивов, не работают, если в образце содержится менее 25–30% опухолевых/аномальных клеток. Однако в онкологических приложениях это ограничение можно свести к минимуму с помощью стратегий обогащения опухолей и программного обеспечения, оптимизированного для использования с онкологическими образцами. Алгоритмы анализа быстро развиваются, и некоторые из них даже предназначены для работы при «обычном заражении клонами». [69] поэтому ожидается, что это ограничение будет продолжать исчезать.
^ Исикава С; Комура Д; Цудзи С; Нисимура К; Ямамото С; Панда Б; Хуан Дж; Фукаяма М; Джонс К.В.; Абуратани Х (август 2005 г.). «Аллельный дозировочный анализ с использованием микрочипов генотипирования». Биохимия Биофиз Рес Коммьюнити . 333 (4): 1309–14. дои : 10.1016/j.bbrc.2005.06.040 . ПМИД 15982637 .
^ Jump up to: а б Ло К.С., Бейли Д., Буркхардт Т., Гардина П., Турпас Ю., Коуэлл Дж. (март 2008 г.). «Комплексный анализ событий потери гетерозиготности при глиобластоме с использованием массивов картирования SNP 100 тыс. и сравнение с аномалиями числа копий, определенными с помощью сравнительной геномной гибридизации массива BAC». Гены Хромосомы Рак . 47 (3): 221–37. дои : 10.1002/gcc.20524 . ПМИД 18050302 . S2CID 19480318 .
^ Михельс Э., Вандесомпель Дж., Хобек Дж., Ментен Б., Де Претер К., Лорейс Г., Ван Рой Н., Спелеман Ф. (2006). «Полногеномное измерение изменений количества копий ДНК при нейробластоме: рассечение ампликонов и картирование потерь, приростов и точек останова». Цитогенет. Геном Рез . 115 (3–4): 273–282. дои : 10.1159/000095924 . ПМИД 17124410 . S2CID 14012430 .
^ Мессахель Б; Уильямс Р.; Ридольфи А; А'херн Р; Уоррен В; Тинворт Л; Хобсон Р; Аль-Саади Р; Уайман Дж; Брандлер М.А.; Келси А; Себире Н; Джонс С; Вуянич Г; Причард-Джонс К.; Группа детского рака и лейкемии (CCLG) (март 2009 г.). «Группа детского рака и лейкемии (CCLG). Потеря аллеля в 16q определяет худший прогноз опухоли Вильмса независимо от подхода к лечению в клинических исследованиях UKW1-3: исследование группы детского рака и лейкемии (CCLG)». Eur J Рак . 45 (5): 819–26. дои : 10.1016/j.ejca.2009.01.005 . ПМИД 19231157 .
^ Лэйгль-Донадей Ф., Бенуаиш-Амиэль А., Хоанг-Сюань К., Сансон М. (2005). «[Молекулярная биология олигодендроглиальных опухолей]». Нейро-хирургия (на французском языке). 51 (3–4, часть 2): 260–8. дои : 10.1016/s0028-3770(05)83487-3 . ПМИД 16292170 .
^ Jump up to: а б Ленц Х.Дж., «Установленные биомаркеры колоректальной карциномы», Учебная книга Американского общества клинической онкологии, 2009, стр. 215-219.
Arc.Ask3.Ru Номер скриншота №: 80977ad503defc955e045534f163a66a__1722208620 URL1:https://arc.ask3.ru/arc/aa/80/6a/80977ad503defc955e045534f163a66a.html Заголовок, (Title) документа по адресу, URL1: Virtual karyotype - Wikipedia
Данный printscreen веб страницы (снимок веб страницы, скриншот веб страницы), визуально-программная копия документа расположенного по адресу URL1 и сохраненная в файл, имеет: квалифицированную, усовершенствованную (подтверждены: метки времени, валидность сертификата), открепленную ЭЦП (приложена к данному файлу), что может быть использовано для подтверждения содержания и факта существования документа в этот момент времени. Права на данный скриншот принадлежат администрации Ask3.ru, использование в качестве доказательства только с письменного разрешения правообладателя скриншота. Администрация Ask3.ru не несет ответственности за информацию размещенную на данном скриншоте. Права на прочие зарегистрированные элементы любого права, изображенные на снимках принадлежат их владельцам. Качество перевода предоставляется как есть. Любые претензии, иски не могут быть предъявлены. Если вы не согласны с любым пунктом перечисленным выше, вы не можете использовать данный сайт и информация размещенную на нем (сайте/странице), немедленно покиньте данный сайт. В случае нарушения любого пункта перечисленного выше, штраф 55! (Пятьдесят пять факториал, Денежную единицу (имеющую самостоятельную стоимость) можете выбрать самостоятельно, выплаичвается товарами в течение 7 дней с момента нарушения.)