Открытие и разработка антиандрогенов
Первый антиандроген был открыт в 1960-х годах. Антиандрогены противодействуют рецептору андрогенов (АР) и тем самым блокируют биологические эффекты тестостерона и дигидротестостерона (ДГТ). Антиандрогены важны для мужчин с гормонально-зависимыми заболеваниями, такими как рак простаты , доброкачественная гиперплазия предстательной железы (ДГП), акне , себорея , гирсутизм и андрогенная алопеция . Антиандрогены в основном используются для лечения заболеваний предстательной железы. [ 1 ] [ 2 ] [ 3 ] Исследования 2010 года показывают, что АР могут быть связаны с прогрессированием трижды негативного рака молочной железы и рака слюнных протоков. [ 4 ] и что антиандрогены потенциально могут быть использованы для его лечения. [ 5 ] [ 6 ]
По состоянию на 2010 год [update] Антиандрогены представляют собой небольшие молекулы и могут быть стероидными или нестероидными в зависимости от химического состава лигандов . Стероидные антиандрогены имеют сходную стероидную структуру, тогда как нестероидные антиандрогены (НПСА) могут иметь структурно различающиеся фармакофоры . Лишь ограниченное количество соединений доступно для клинического использования, несмотря на то, что было обнаружено и исследовано очень большое разнообразие антиандрогенных соединений. [ 2 ]
История
[ редактировать ]В начале двадцатого века связь между гипофизом , яичками и предстательной железой была установлена . Американский врач Чарльз Брентон Хаггинс обнаружил, что кастрация или введение эстрогена желез приводят к атрофии у мужчин, которую можно обратить вспять повторным введением андрогена. В 1941 году Хаггинс лечил пациентов с раком простаты методом андрогенной абляции с кастрацией или терапией эстрогенами; было осознано благотворное влияние андрогенной абляции на метастатический рак простаты, за что он был удостоен Нобелевской премии по физиологии и медицине в 1966 году. [ 1 ]
Стало очевидным, что одной андрогенной абляции недостаточно для лечения пациентов с распространенным раком простаты. В конце 1960-х годов был открыт и охарактеризован андрогенный рецептор (АР). Скрининг химических библиотек на наличие блокаторов АР привел к открытию первого антиандрогена — ципротерона . группа ацетатная Затем к ципротерону была добавлена и образовался ацетат ципротерона . антиандроген флутамид В 1970-х годах был открыт . США В 1989 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило его использование для лечения рака простаты. В 1995 году был одобрен бикалутамид , а нилутамид . год спустя — [ 1 ] [ 7 ]
Андрогенный рецептор
[ редактировать ]
AR принадлежит к стероидных рецепторов подсемейству суперсемейства ядерных рецепторов . Его функция регулируется связыванием андрогенов , что инициирует последовательные конформационные изменения рецептора , влияющие на взаимодействия рецептор- белок и рецептор-ДНК. Эндогенными андрогенами являются в основном тестостерон и ДГТ. [ 8 ] [ 9 ] [ 10 ] [ 11 ] АР экспрессируется в клетках широкого спектра тканей по всему организму, за пределами первичных и вторичных половых органов. [ 12 ]
AR Ген имеет длину более 90 КБ и кодирует белок, состоящий из 919 аминокислот . У человека идентифицирован только один ген AR, который расположен на хромосоме X. Он состоит из четырех основных регионов, см. рисунок 1: [ 2 ] [ 3 ] [ 7 ] [ 8 ]
- N-концевой домен (NTD), выполняющий модулирующую функцию.
- ДНК-связывающий домен (DBD), который распознает и связывается с элементами ответа на андрогены (ARE) в последовательности целевого гена.
- Лигандсвязывающий домен (LBD), отвечающий за распознавание и связывание лиганда.
- Небольшая шарнирная область между DBD и LBD.
В AR были идентифицированы две функции, которые играют решающую роль в регуляции трансактивации гена-мишени : N-концевая функция активации 1 (AF1) и C-концевая функция активации 2 (AF2). AF1 не зависит от лиганда и играет основную роль в трансактивации гена-мишени. AF2 является лиганд-зависимым и выполняет лишь ограниченную функцию. [ 8 ] [ 10 ]
Механизм действия
[ редактировать ]Несвязанный AR в основном расположен в цитоплазме , как типичный стероидный рецептор, и связан с комплексом белков теплового шока (HSP) посредством взаимодействия с LBD. Андрогены, как агонисты , так и антагонисты, располагаются в лиганд-связывающем кармане (LBP) цитозольного AR и связываются с LBD, см. рисунок 2. AR претерпевает ряд конформационных изменений, и HSP диссоциирует от AR. Трансформированный AR подвергается димеризации , фосфорилированию и транспортируется в ядро. Транслоцированный рецептор затем связывается с элементами андрогенного ответа (ARE) на промоторе андроген-чувствительного гена, консенсусной последовательности, расположенной либо выше, либо ниже сайта начала транскрипции (TSS) генов-мишеней AR. Привлечение других кофакторов транскрипции (включая коактиваторы и корепрессоры) и общего механизма транскрипции дополнительно обеспечивает трансактивацию экспрессии генов , регулируемой AR . Все эти сложные процессы инициируются лиганд-индуцированными конформационными изменениями в LBD. Лиганд-специфическое рекрутирование корегуляторы могут иметь решающее значение для агонистической или антагонистической активности лигандов AR. Связывание ДНК также необходимо для экспрессии генов, регулируемой AR, также известной как классическая функция геномного гена AR. [ 7 ] [ 8 ] [ 10 ]
Разработка антиандрогенов
[ редактировать ]Ципротерон представляет собой стероидный антиандроген, который конкурентно ингибирует связывание тестостерона или ДГТ с АР. Ципротерон связывается с AR, которые экспрессируются клетками рака простаты, а также с AR, которые экспрессируются в гипоталамусе и гипофизе. Таким образом, ципротерон блокирует отрицательную обратную связь андрогенов на гипоталамо-гипофизарном уровне, что приводит к повышению уровня лютеинизирующего гормона (ЛГ) в сыворотке. Это повышение уровня ЛГ вызывает повышение уровня тестостерона в сыворотке крови и в конечном итоге снижает способность ципротерона конкурировать за связывание АР и блокировать андрогенную стимуляцию . [ 1 ] [ 7 ]
Ципротерона ацетат был разработан для решения этой проблемы. Он образуется путем добавления ацетатной группы к ципротерону, см. рисунок 3. Ацетат ципротерона имеет двойной механизм действия, поскольку он напрямую конкурирует с ДГТ за связывание с АР, но также ингибирует секрецию гонадотропинов . Тем самым он снижает уровни андрогенов, эстрогенов и ЛГ. [ 1 ] [ 7 ] Ципротерона ацетат действует как непосредственно как антиандроген в клетках рака простаты, так и косвенно снижает уровень тестостерона в сыворотке. Последнее вызывает ограничения ципротерона ацетата, которые оказывают центральное воздействие на секрецию андрогенов, с последующей потерей либидо и сексуальной потенции. В нескольких сообщениях также утверждается, что ципротерона ацетат вызывает гиперплазию печени . Эти побочные эффекты дали фармацевтическим компаниям стимул искать альтернативные, «чистые» НПВП, которые не имели бы этих побочных эффектов. [ 1 ] Чистые антиандрогены блокируют рецепторы андрогенов, не оказывая агонистической или какой-либо другой гормональной активности. [ 3 ]
Флутамид стал первым НПВП, прошедшим клинические испытания. Позже были разработаны НПВП бикалутамид и нилутамид. Предполагаемые преимущества этих соединений заключались в том, что они не влияли на либидо или потенцию, как другие разрабатываемые соединения центрального действия, агонисты рилизинг-гормона лютеинизирующего гормона (LHRH) и ацетат ципротерона. Но эта теория не подтвердилась. Эти НПСА в конечном итоге проникли через гематоэнцефалический барьер , как и ацетат ципротерона, что привело к последующему увеличению уровня тестостерона в сыворотке. [ 1 ]
Флутамид
[ редактировать ]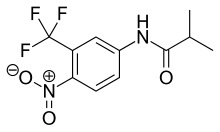
арилпропионамида Флутамид представляет собой аналог с чистыми антиандрогенными свойствами, как показано на рисунке 4. Он полностью всасывается из желудочно-кишечного тракта после перорального приема и подвергается интенсивному метаболизму при первом прохождении до его активной формы, 2-гидроксифлутамида, и продукта гидролиза , 3-трифторметил-4. -нитроанилин. [ 7 ] [ 9 ] [ 10 ] Гидроксифлутамид является более сильным антагонистом AR, чем флутамид in vivo , с более высокой аффинностью связывания с AR. гидроксифлутамида Период полувыведения у человека составляет около 8 часов. Гидролиз амидной связи представляет собой основной путь метаболизма этого активного метаболита . Обращая вспять стимулирующий эффект ДГТ на вес вентральной части простаты, флутамид примерно в 2 раза более эффективен , чем ацетат ципротерона. Гидроксифлутамид имеет относительно низкую аффинность связывания с АР и поэтому обычно используется в высоких дозах для достижения полной блокировки АР в терапии. [ 9 ] [ 13 ]
Нилутамид
[ редактировать ]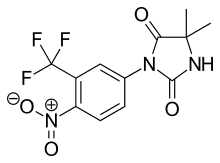
Нилутамид представляет собой нитроароматический гидантоиновый аналог флутамида, как показано на рисунке 5. [ 9 ] [ 10 ] Нилутамид выводится исключительно путем метаболизма, преимущественно за счет восстановления ароматической нитрогруппы . Хотя был выявлен гидролиз одной из карбонильных функций имидазолиндиона, она гораздо менее подвержена печеночному метаболизму, чем амидная связь в гидроксифлутамиде. Это приводит к увеличению периода полувыведения нилутамида у людей, составляющему 2 дня. Тем не менее, нитроанионный свободный радикал, образующийся при восстановлении нитро, все еще может быть связан с гепатотоксичностью у людей, особенно при использовании относительно высоких доз, применяемых для блокирования андрогенов. [ 9 ] Нилутамид вызывает побочные эффекты, которые ограничивают его использование, такие как пневмонит и задержка адаптации к темноте. [ 7 ]
Бикалутамид
[ редактировать ]
Бикалутамид представляет собой аналог арилпропионамида, показанный на рисунке 6. [ 9 ] [ 10 ] Он заменил флутамид и нилутамид в качестве антиандрогена первого выбора для лечения рака простаты. Бикалутамид не так гепатотоксичен, как флутамид и нилутамид, и имеет более длительный период полувыведения - 6 дней у человека, что позволяет назначать его один раз в день в более низких дозах. Бикалутамид разделяет структуру амидной связи с флутамидом. Несмотря на это, гидролиз амидной связи был обнаружен у крыс, а не у людей, что может объяснить длительный период полураспада бикалутамида у людей. [ 9 ]
Бикалутамид имеет цианогруппу в пара-положении вместо нитрогруппы, как у флутамида и нилутамида. Такое изменение групп позволяет избежать нитровосстановления, наблюдаемого в нилутамиде. Бикалутамид имеет в своей структуре хиральный углерод (помечен звездочкой на рисунке 6), который связан с гидроксильной и метильной группами. Поэтому его применяют в виде рацемата . [ 9 ] Исследование, проведенное после регистрации препарата, показало, что его антиандрогенная активность почти полностью связана с (R) -энантиомером . (R)-бикалутамид имеет почти в четыре раза более высокое сродство к АР предстательной железы, чем гидроксифлутамид, и имеет лучший профиль побочных эффектов по сравнению с другими антиандрогенами. [ 9 ] [ 10 ]
Связь структуры и деятельности
[ редактировать ]Стероидные антиандрогены
[ редактировать ]Ципротерона ацетат представляет собой 6-хлор-1,2-метиленовое производное 17α-ацетоксипрогестерона. Он проявляет основную антиандрогенную активность наряду с андрогенной активностью. Ципротерона ацетат проявляет высокое сродство к AR у крыс, которое увеличивается при удалении 1,2-метиленовой группы из соединения. Если атом хлора заменяется метильной группой, связывание несколько уменьшается, тогда как дальнейшее удаление двойной связи C6 изменяет кинетику связывания, см. рисунок 7. [ 3 ]

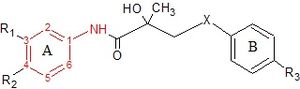

Нестероидные антиандрогены
[ редактировать ]Гидроксифлутамид и его аналоги, бикалутамид и нилутамид, имеют анилидную кольцевую структуру. Структуры можно увидеть на рисунке 7, где анилидное кольцо окрашено в красный цвет. Этим трем соединениям требуется ароматическое кольцо с дефицитом электронов для эффективного связывания AR. Замена анилида алкеном приводит к образованию слабоактивных соединений, что можно объяснить отсутствием внутримолекулярного связывания водорода или плохой способностью донора водородных связей. [ 3 ] Различные комбинации электроноакцепторных замен в анилиновом кольце этих препаратов не показали более высокого связывания с AR-рецептором по сравнению с соединениями, имеющими хлор- или трифторметильную группу в мета-положении (R1) и либо циано-, либо азотистую группу в мета-положении (R1) и либо циано-, либо азотистую группу в мета-положении. пара-позиция (R2). [ 3 ] [ 14 ]
Для гидроксифлутамида группа соединений, различающихся ароматическим кольцом, не связывалась с АР. Это предполагает, что бизамещение в гидроксифлутамидном кольце важно для высокой аффинности связывания AR. Также было продемонстрировано, что гидроксифлутамид требует сильной донорной способности третичной гидроксильной группы и фиксированных конформеров, участвующих во внутримолекулярном связывании водорода, для эффективного связывания с AR. [ 3 ] [ 14 ]
Для бикалутамида антиандрогенная активность сульфидных и сульфоновых замен Х-связи была протестирована in vitro . Сульфиды в большинстве случаев показали как минимум в 2 раза более высокую аффинность связывания, чем соответствующие сульфоны. Однако эта зависимость изменилась, когда группа R3 представляла собой NHSO 2 CH 3 , где аффинность связывания сульфона была в 3 раза выше, чем у сульфида. Эти результаты указывают на то, что заместители B-кольца во многом определяют эффект X-связи при связывании AR. Исследователи предположили, что третичная гидроксильная группа участвует в прямом взаимодействии с AR, поскольку, когда ацетильная группа, аффинность связывания рецептора значительно снижается. к этой гидроксильной группе вводится [ 14 ]
Нилутамид имеет очень низкое сродство к АР при тестировании на простате кастрированных крыс. Модификации, такие как замена атома N3 на кислород, мало влияют на сродство соединения к АР предстательной железы. При замене атома кислорода на атом серы в положении С2 имидазольного кольца и добавлении бутилового спирта к атому N3 рецепторсвязывающая и биологическая активность соединения увеличивается в 100 раз по сравнению с НПАК. Также соединение не связывается с другими стероидными рецепторами. Если метильную группу заменить на группу бутилового спирта, соединение проявит в 3 и 10 раз большую антиандрогенную активность in vivo , чем бикалутамид и нилутамид соответственно. [ 3 ]
Синдром отмены антиандрогенов
[ редактировать ]Антиандрогены, которые в настоящее время доступны на рынке, особенно полезны для лечения рака простаты на ранних стадиях. Однако рак предстательной железы часто прогрессирует до гормонорефрактерного состояния, при котором рак прогрессирует на фоне продолжающейся андрогенной абляции или антиандрогенной терапии. [ 9 ] Это говорит о том, что длительное использование этих антиандрогенов при раке простаты может привести к развитию андроген-независимых клеток рака простаты или способности андрогенов надпочечников поддерживать рост опухоли . [ 8 ] Это явление называется синдромом отмены антиандрогенов (САМ) и является одним из основных недостатков существующих антиандрогенов. AWS определяется как регрессия опухоли или облегчение симптомов, наблюдаемое после прекращения антиандрогенной терапии. Механизм этого до конца не ясен, но современные теории включают изменения гена AR, белков-корегуляторов и/или путей передачи сигнала. Эта резистентность к антиандрогенам также может быть связана с относительной слабостью современных антиандрогенов, поскольку их сродство к АР в 50 и более раз ниже, чем у ДГТ. Это также может объяснить, почему часто наблюдается компенсаторная сверхэкспрессия AR. [ 7 ]
Мутации гена рецептора андрогена
[ редактировать ]гена AR Мутации в LBD, которые изменяют специфичность и/или функциональную активность лиганда, существуют и, как полагают, способствуют превращению некоторых антагонистов AR в агонисты, что объясняет парадоксальное временное улучшение, иногда наблюдаемое у пациентов после прекращения антиандрогенной терапии. [ 15 ] Эти мутации могут иметь большое влияние на антагонистическую активность нынешних низкомолекулярных антиандрогенов и делать их менее эффективными в блокировании функции AR посредством непрямой модуляции изнутри LBP. Недавние исследования циркулирующих опухолевых клеток показывают, что частота мутаций выше, чем предполагалось ранее на основании биопсии опухолей . [ 16 ] Т877А , [ 17 ] W741L и W741C Мутации [ 18 ] являются примерами известных мутаций AR LBD. Линия LNCaP рака простаты клеток экспрессирует AR с точечной мутацией T877A, которая вызывает пролиферацию в присутствии антиандрогенов гидроксифлутамида и ацетата ципротерона. Эта мутация также была обнаружена у пациентов с синдромом отмены антиандрогенов, получающих лечение этими соединениями. [ 17 ] В другом исследовании обработка клеток LNCaP бикалутамидом привела к двум мутациям LBD: W741L и W741C. [ 18 ] заставляя бикалутамид приобретать агонистическую активность в отношении обоих мутантных AR. [ 19 ] Мутация W741L создает дополнительное пространство, так что сульфонильное связанное фенильное кольцо бикалутамида размещается на месте отсутствующего индольного кольца W741. [ 20 ] В немутантном AR наличие боковой цепи W741, вероятно, заставляет бикалутамид выступать наружу, что предотвращает активное положение H12 на рецепторе AR. Однако гидроксифлутамид работал как антагонист мутантных AR W741. [ 18 ] Это согласуется с теорией о том, что флутамид и нилутамид противодействуют АР по механизму «пассивного антагонизма», поскольку они имеют более скромные размеры, чем бикалутамид. [ 20 ] Таким образом, эти препараты могут быть эффективны в качестве терапии второй линии при рефрактерном раке предстательной железы, ранее лечившемся бикалутамидом. [ 18 ]
Текущий статус
[ редактировать ]Антагонисты N-концевого домена
[ редактировать ]Было предложено, что антагонисты N-концевого домена (NTD) AR преодолевают ограничения современных антиандрогенов в отношении мутантных AR путем прямого блокирования функции AR с поверхности белка, за пределами LBP. Считается, что эта прямая блокада обеспечивает более эффективную стратегию предотвращения или преодоления аномального действия AR во время AWS, а также обеспечивает большую гибкость в структурных модификациях без ограничений по пространству, присущих жесткой LBP. [ 8 ]
Стероидные рецепторы имеют сходство в последовательностях генов и белковых структурах, что часто приводит к функциональному перекрестному взаимодействию между стероидными рецепторами. Одним из критериев выбора антагонистов AR NTD является достижение высокой степени специфичности к AR. Однако важно понимать, что специфичность AR не обязательно транслируется in vivo , поскольку антагонисты NTD могут также взаимодействовать с белками-мишенями, отличными от AR. [ 8 ]
Лигандсвязывающий домен как целевой сайт
[ редактировать ]Активация AR требует образования области функциональной функции активации 2 (AF2) в AR LBD, которая опосредует взаимодействия между AR и различными кофакторами транскрипции . Таким образом, большая часть исследований антагонистов NTD AR сосредоточена на пептидах , которые могут напрямую блокировать AF2 в LBD AR с поверхности белка. Даже в связанном мутантном AR антагонисты NTD будут способны блокировать функцию AF2 посредством прямого поверхностного взаимодействия, независимо от связанного лиганда. [ 8 ]
Исследования этих антагонистов NTD обычно проводятся путем аффинного скрининга библиотек фагового дисплея , которые экспрессируют случайные пептиды, содержащие различные сигнатурные мотивы . AR, по-видимому, отдают явное предпочтение мотивам связывания типа «FxxLF» (где F = фенилаланин , L = лейцин и X = любой аминокислотный остаток), тогда как другие ядерные рецепторы имеют очень похожий механизм связывания для типа «LxxLL». связующие мотивы. Это открывает уникальную возможность для разработки AR-специфичных пептидов. [ 8 ]
Несмотря на то, что низкомолекулярные антагонисты и антагонисты NTD, нацеленные на поверхность AF2, различаются сайтами связывания, они оба ингибируют функцию AR, нарушая функцию AF2. Следовательно, с механистической точки зрения эти антагонисты NTD также могут быть классифицированы как «антагонисты AF2». [ 8 ]
Домен N-Terminal в качестве целевого сайта
[ редактировать ]Функционально AR NTD играет основную роль в регуляции активации транскрипции гена-мишени и опосредовании различных рецептор-белковых и внутрирецепторных N-концевых и C-концевых взаимодействий. Таким образом, модуляция функции NTD считается эффективной стратегией воздействия на AR. Среди различных функциональных доменов в различных ядерных рецепторах NTD является наименее консервативным и поэтому может стать лучшим местом-мишенью для антагонистов NTD для достижения AR-специфичности. Однако структурные особенности NTD не определены из-за высокой степени гибкости его конформации. Как биохимический анализ , так и спектроскопический анализ кругового дихроизма показывают, что AR NTD сильно разупорядочен в нативных условиях, что делает его трудной мишенью для поиска лекарств. [ 8 ]
В 2008 году появились сообщения о хлорированном пептиде синтокамиде А , выделенном из морских губок, который эффективно ингибирует транскрипцию репортерного гена, активируемого N-концевым доменом AR, см. рисунок 8. [ 21 ] Представленных доказательств было недостаточно, чтобы поддержать вывод о том, что синтокамид А напрямую ингибирует функцию AR NTD, и механизм действия требует дальнейшего изучения. [ 8 ]
Селективные модуляторы андрогенных рецепторов
[ редактировать ]Доступные сегодня низкомолекулярные антиандрогены обладают нежелательными побочными эффектами, вызванными полным, неселективным ингибированием действия АР. новый класс тканевых селективных модуляторов андрогенных рецепторов Чтобы свести к минимуму эти побочные эффекты, в качестве нового подхода к лечению рака простаты был предложен (SARM). Эти лиганды должны вести себя как антагонисты в предстательной железе, либо не проявляя активности, либо не проявляя активности агониста в других тканях-мишенях, чтобы оказывать незначительное воздействие или вообще не оказывать никакого воздействия на анаболические ткани или центральную нервную систему (ЦНС). Однако открытие этого нового класса лигандов может оказаться сложной задачей, поскольку молекулярный механизм действия AR недостаточно изучен. [ 8 ]
Было предложено несколько механизмов для достижения тканевой селективности лигандов AR. Существуют наиболее убедительные доказательства роли 5-альфа-редуктазы . 5-альфа-редуктаза экспрессируется только в определенных тканях и поэтому может вносить уникальный вклад в тканевую селективность. Специфическое ингибирование фермента типа 2 финастеридом блокирует превращение тестостерона в ДГТ в простате. [ 8 ]
Несколько подходов могут использовать потенциальную тканеспецифическую конверсию для разработки SARM, в том числе:
- Неактивные исходные соединения, которые активируются 5-альфа-редуктазой 2 типа в предстательной железе с образованием антиандрогенов.
- Агонисты AR, которые инактивируются 5-альфа-редуктазой 2 типа в простате.
- Агонисты AR, которые превращаются в антиандрогены только под действием 5-альфа-редуктазы 2 типа в предстательной железе. [ 22 ]
Другие низкомолекулярные антиандрогены
[ редактировать ]Статус разработки других низкомолекулярных антиандрогенов, исследование которых проводилось в 2011 году, можно увидеть в таблице 1.
| Название соединения | Структура | Компания | Стадия разработки | Другая информация | |
|---|---|---|---|---|---|
| RU58642 |  |
Руссель-Уклаф СА | Доклинический – никаких дальнейших разработок с 1998 г. | Перорально активен и более эффективен, чем современные низкомолекулярные антиандрогены . [ 23 ] | |
| LG120907 | 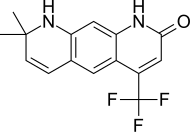 |
Лиганд Фармасьютикалс | Доклинический | Активен при пероральном приеме, оказывает сильное антагонистическое действие на предстательную железу без повышения уровня ЛГ и тестостерона в плазме . [ 24 ] | |
| LG105 | 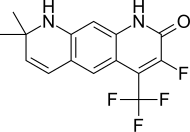 |
Лиганд Фармасьютикалс | Доклинический | При пероральном приеме оказывает сильное антагонистическое действие на простату без повышения уровня ЛГ и тестостерона в плазме. Кажется, более мощный, чем LG120907. [ 24 ] | |
| Апалутамид (Эрлеада) |  |
Медитация | Одобренный | Высокая аффинность связывания с AR. В отличие от бикалутамида , он не способствует ядерной транслокации и ухудшает как связывание ДНК с элементами андрогенного ответа, так и рекрутирование коактиваторов . [ 25 ] | |
| Энзалутамид (Кстанди) |  |
Медитация | Одобренный | Высокая аффинность связывания с AR. В отличие от бикалутамида, он не способствует ядерной транслокации и ухудшает как связывание ДНК с элементами андрогенного ответа, так и рекрутирование коактиваторов. [ 25 ] Индуцирует апоптоз опухолевых клеток и не обладает агонистической активностью. [ 26 ] | |
| БМС-641988 | 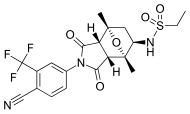 |
Бристол-Майерс Сквибб | Клиническая фаза I – исследование прекращено | Показал повышенную эффективность по сравнению с бикалутамидом. Исследование фазы I было прекращено из-за эпилептического припадка у пациента. [ 27 ] Привели к выводу, что некоторые антиандрогены вызывают нецелевое антагонистическое связывание с рецепторами ГАМК-А . [ 28 ] | |
| CH5137291 | 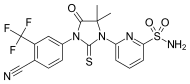 |
Чугай Фармасьютикал Ко. Лтд. | Доклинический | Полностью ингибирует AR-опосредованную трансактивацию и пролиферацию модели ксенотрансплантата CRPC LNCaP-BC2, устойчивой к бикалутамиду. [ 29 ] [ 30 ] |
 |
 |
Природные антиандрогены
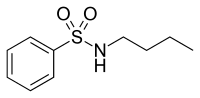
[ редактировать ]Атрариновая кислота и N-бутилбензолсульфонамид — природные соединения с антиандрогенными свойствами, полученные из коры африканского дерева Pygeum africanum , см. рисунки 9 и 10. [ 31 ] Анализы in vitro показали, что они оба являются селективными агонистами AR и ингибируют пролиферацию нескольких линий клеток рака простаты. Атрариновая кислота также препятствует инвазии внеклеточного матрикса, и оба соединения способны предотвращать андроген-индуцированную ядерную транслокацию AR. В настоящее время синтезируются более мощные производные в надежде улучшить фармакологический профиль этих двух соединений. [ 32 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г Денмид С.Р., Айзекс Дж.Т. (май 2002 г.). «История лечения рака простаты» . Обзоры природы. Рак . 2 (5): 389–96. дои : 10.1038/nrc801 . ПМЦ 4124639 . ПМИД 12044015 .
- ^ Jump up to: а б с Гао В. (октябрь 2010 г.). «Андрогеновый рецептор как терапевтическая мишень». Обзоры расширенной доставки лекарств . 62 (13): 1277–84. дои : 10.1016/j.addr.2010.08.002 . ПМИД 20708648 .
- ^ Jump up to: а б с д и ж г час Сингх С.М., Готье С., Лабри Ф. (февраль 2000 г.). «Антагонисты андрогенных рецепторов (антиандрогены): взаимосвязь структура-активность». Современная медицинская химия . 7 (2): 211–47. дои : 10.2174/0929867003375371 . ПМИД 10637363 .
- ^ Уильямс Л., Томпсон Л.Д., Ситхала Р.Р., Вайнреб И., Ассаад А.М., Тулук М. и др. (май 2015 г.). «Рак слюнных протоков: преобладание апокринной морфологии, преобладание гистологических вариантов и экспрессия андрогенных рецепторов». Американский журнал хирургической патологии . 39 (5): 705–13. doi : 10.1097/pas.0000000000000413 . ПМИД 25871467 . S2CID 24737257 .
- ^ Гукальп А., Трейна Т.А. (январь – февраль 2010 г.). «Тройной негативный рак молочной железы: роль андрогенного рецептора». Раковый журнал . 16 (1): 62–5. дои : 10.1097/PPO.0b013e3181ce4ae1 . ПМИД 20164692 . S2CID 6922842 .
- ^ Урбан Д., Ришин Д., Анхель С., Д'Коста И., Соломон Б. (март 2015 г.). «Абиратерон при метастатическом раке слюнных протоков» . Журнал Национальной комплексной онкологической сети . 13 (3): 288–90. дои : 10.6004/jnccn.2015.0040 . ПМИД 25736005 .
- ^ Jump up to: а б с д и ж г час Хендлер Б., Клив А. (апрель 2012 г.). «Последние разработки в области антиандрогенов и селективных модуляторов андрогенных рецепторов». Молекулярная и клеточная эндокринология . 352 (1–2): 79–91. дои : 10.1016/j.mce.2011.06.002 . ПМИД 21704118 . S2CID 36184991 .
- ^ Jump up to: а б с д и ж г час я дж к л м н Гао В (2010). «Пептидный антагонист рецептора андрогенов». Текущий фармацевтический дизайн . 16 (9): 1106–13. дои : 10.2174/138161210790963850 . ПМИД 20030610 .
- ^ Jump up to: а б с д и ж г час я дж Гао В., Ким Дж., Далтон Дж.Т. (август 2006 г.). «Фармакокинетика и фармакодинамика лигандов нестероидных рецепторов андрогенов» . Фармацевтические исследования . 23 (8): 1641–58. дои : 10.1007/s11095-006-9024-3 . ПМК 2072875 . ПМИД 16841196 .
- ^ Jump up to: а б с д и ж г Лемке Т.Л., Уильямс Д.А. (2002). Принципы медицинской химии Фоя (5-е изд.). Балтимор [и др.]: Уильямс и Уилкинс. ISBN 978-0-683-30737-5 .
- ^ Нарайанан Р., Молер М.Л., Бол С.Э., Миллер Д.Д., Далтон Дж.Т. (2008). «Селективные модуляторы андрогенных рецепторов в доклинической и клинической разработке» . Передача сигналов ядерных рецепторов . 6 : e010. дои : 10.1621/nrs.06010 . ПМК 2602589 . ПМИД 19079612 .
- ^ Гельманн Е.П. (июль 2002 г.). «Молекулярная биология рецептора андрогенов». Журнал клинической онкологии . 20 (13): 3001–15. дои : 10.1200/jco.2002.10.018 . ПМИД 12089231 .
- ^ Пойет П., Лабри Ф. (октябрь 1985 г.). «Сравнение антиандрогенной/андрогенной активности флутамида, ацетата ципротерона и ацетата мегестрола». Молекулярная и клеточная эндокринология . 42 (3): 283–8. дои : 10.1016/0303-7207(85)90059-0 . ПМИД 3930312 . S2CID 24746807 .
- ^ Jump up to: а б с Инь Д., Хе Ю., Перера М.А., Хонг С.С., Мархефка С., Стурман Н. и др. (январь 2003 г.). «Ключевые структурные особенности нестероидных лигандов для связывания и активации рецептора андрогена» . Молекулярная фармакология . 63 (1): 211–23. дои : 10.1124/моль.63.1.211 . ПМК 2040236 . ПМИД 12488554 .
- ^ Миямото Х., Рахман М.М., Чанг С. (январь 2004 г.). «Молекулярные основы синдрома отмены антиандрогенов». Журнал клеточной биохимии . 91 (1): 3–12. дои : 10.1002/jcb.10757 . ПМИД 14689576 . S2CID 5773128 .
- ^ Цзян Ю, Пальма Дж. Ф., Агус Д. Б., Ван Ю, Гросс МЭ (сентябрь 2010 г.). «Обнаружение мутаций андрогенных рецепторов в циркулирующих опухолевых клетках при кастрационно-резистентном раке простаты» (PDF) . Клиническая химия . 56 (9): 1492–5. дои : 10.1373/clinchem.2010.143297 . ПМИД 20581083 . Архивировано (PDF) из оригинала 17 августа 2017 г. Проверено 2 сентября 2019 г.
- ^ Jump up to: а б Сузуки Х, Акакура К, Комия А, Аида С, Акимото С, Симадзаки Дж (сентябрь 1996 г.). «Мутация кодона 877 в гене рецептора андрогенов при распространенном раке простаты: связь с синдромом отмены антиандрогенов». Простата . 29 (3): 153–8. doi : 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5 . ПМИД 8827083 . S2CID 22806905 .
- ^ Jump up to: а б с д Хара Т., Миядзаки Дж., Араки Х., Ямаока М., Канзаки Н., Кусака М. и др. (январь 2003 г.). «Новые мутации андрогенных рецепторов: возможный механизм синдрома отмены бикалутамида». Исследования рака . 63 (1): 149–53. ПМИД 12517791 .
- ^ Бол С.Э., Гао В., Миллер Д.Д., Белл С.Э., Далтон Дж.Т. (апрель 2005 г.). «Структурные основы антагонизма и резистентности бикалутамида при раке простаты» . Труды Национальной академии наук Соединенных Штатов Америки . 102 (17): 6201–6. Бибкод : 2005PNAS..102.6201B . дои : 10.1073/pnas.0500381102 . ПМЦ 1087923 . ПМИД 15833816 .
- ^ Jump up to: а б Нале З. (2008). «Функциональный и структурный анализ андрогенных рецепторов для открытия противораковых лекарств» (PDF) . Терапия рака . 6 : 439–444. Архивировано из оригинала (PDF) 24 апреля 2012 г. Проверено 27 сентября 2011 г.
- ^ Садар М.Д., Уильямс Д.Э., Мауджи Н.Р., Патрик Б.О., Виканта Т., Часана Э. и др. (ноябрь 2008 г.). «Синтокамиды от A до E, хлорированные пептиды из губки Dysidea sp., которые ингибируют трансактивацию N-конца рецептора андрогена в клетках рака простаты». Органические письма . 10 (21): 4947–50. дои : 10.1021/ol802021w . ПМИД 18834139 .
- ^ Гао В., Далтон Дж. Т. (март 2007 г.). «Расширение терапевтического использования андрогенов с помощью селективных модуляторов андрогенных рецепторов (SARM)» . Открытие наркотиков сегодня . 12 (5–6): 241–8. дои : 10.1016/j.drudis.2007.01.003 . ПМК 2072879 . ПМИД 17331889 .
- ^ Баттманн Т., Бранш С., Бушу Ф., Середе Э., Филибер Д., Губе Ф. и др. (январь 1998 г.). «Фармакологический профиль RU 58642 — мощного системного антиандрогена для лечения андрогензависимых заболеваний». Журнал биохимии стероидов и молекулярной биологии . 64 (1–2): 103–11. дои : 10.1016/S0960-0760(97)00151-9 . ПМИД 9569015 . S2CID 290926 .
- ^ Jump up to: а б Хаманн Л.Г., Хигучи Р.И., Чжи Л., Эдвардс Дж.П., Ван С.Н., Маршке К.Б. и др. (февраль 1998 г.). «Синтез и биологическая активность новой серии нестероидных периферически селективных антагонистов андрогенных рецепторов, полученных из 1,2-дигидропиридоно[5,6-г]хинолинов». Журнал медицинской химии . 41 (4): 623–39. дои : 10.1021/jm970699s . ПМИД 9484511 .
- ^ Jump up to: а б Тран С., Оук С., Клегг Н.Дж., Чен Ю., Уотсон П.А., Арора В. и др. (май 2009 г.). «Разработка антиандрогена второго поколения для лечения распространенного рака простаты» . Наука . 324 (5928): 787–90. Бибкод : 2009Sci...324..787T . дои : 10.1126/science.1168175 . ПМК 2981508 . ПМИД 19359544 .
- ^ Шер Х.И., Бир Т.М., Хигано К.С., Ананд А., Таплин М.Е., Эфстатиу Э. и др. (апрель 2010 г.). «Противоопухолевая активность MDV3100 при кастрационно-резистентном раке простаты: исследование фазы 1-2» . Ланцет . 375 (9724): 1437–46. дои : 10.1016/S0140-6736(10)60172-9 . ПМЦ 2948179 . ПМИД 20398925 .
- ^ Раткопф Д., Лю Г., Кардуччи М.А., Айзенбергер М.А., Ананд А., Моррис М.Дж. и др. (февраль 2011 г.). «Исследование фазы I с увеличением дозы нового антиандрогена BMS-641988 у пациентов с кастрационно-резистентным раком простаты» . Клинические исследования рака . 17 (4): 880–7. дои : 10.1158/1078-0432.CCR-10-2955 . ПМК 3070382 . ПМИД 21131556 .
- ^ Фостер В.Р., Кар Б.Д., Ши Х., Левеск П.С., Обермайер М.Т., Ган Дж. и др. (апрель 2011 г.). «Безопасность лекарств является препятствием для открытия и разработки новых антагонистов андрогенных рецепторов». Простата . 71 (5): 480–8. дои : 10.1002/pros.21263 . ПМИД 20878947 . S2CID 24620044 .
- ^ Кавата Х., Араи С., Накагава Т., Исикура Н., Нишимото А., Ёсино Х. и др. (сентябрь 2011 г.). «Биологические свойства чистого антагониста андрогенных рецепторов для лечения резистентного к кастрации рака простаты: оптимизация от соединения свинца до CH5137291». Простата . 71 (12): 1344–56. дои : 10.1002/pros.21351 . ПМИД 21308717 . S2CID 42009977 .
- ^ Ёсино Х., Сато Х., Сираиси Т., Тачибана К., Эмура Т., Хонма А. и др. (декабрь 2010 г.). «Разработка и синтез чистого антагониста андрогенных рецепторов (CH5137291) для лечения резистентного к кастрации рака простаты». Биоорганическая и медицинская химия . 18 (23): 8150–7. дои : 10.1016/j.bmc.2010.10.023 . ПМИД 21050768 .
- ^ Шляйх С., Папайоанну М., Баниахмад А., Матуш Р. (июль 2006 г.). «Экстракты Pygeum africanum и других этноботанических видов с антиандрогенной активностью». Планта Медика . 72 (9): 807–13. дои : 10.1055/s-2006-946638 . ПМИД 16783690 . S2CID 260278595 .
- ^ Роелл Д., Баниахмад А. (январь 2011 г.). «Природные соединения атрариновой кислоты и N-бутилбензолсульфонамида как антагонисты андрогенных рецепторов человека и ингибиторы роста клеток рака простаты» (PDF) . Молекулярная и клеточная эндокринология . 332 (1–2): 1–8. дои : 10.1016/j.mce.2010.09.013 . ПМИД 20965230 . S2CID 26865620 . Архивировано (PDF) из оригинала 01 октября 2019 г. Проверено 1 октября 2019 г.