Генное предсказание

В вычислительной биологии предсказание генов или поиск генов относится к процессу идентификации областей геномной ДНК, которые кодируют гены . , кодирующие белки, Это включает в себя гены а также гены РНК , но может также включать предсказание других функциональных элементов, таких как регуляторные области . Обнаружение генов является одним из первых и наиболее важных шагов в понимании генома вида после его секвенирования .
На заре своего существования «поиск генов» основывался на кропотливых экспериментах над живыми клетками и организмами. Статистический анализ скорости гомологичной рекомбинации нескольких различных генов мог бы определить их порядок на определенной хромосоме , а информацию из многих таких экспериментов можно было бы объединить для создания генетической карты, определяющей приблизительное расположение известных генов относительно друг друга. Сегодня, когда в распоряжении исследовательского сообщества имеется комплексная последовательность генома и мощные вычислительные ресурсы, поиск генов был переопределен как преимущественно вычислительная задача.
Определение функциональности последовательности следует отличать от определения функции гена или его продукта. Прогнозирование функции гена и подтверждение точности предсказания гена по-прежнему требуют in vivo . экспериментов [1] посредством нокаута генов и других анализов, хотя границы в области биоинформатики исследований [2] делают все более возможным предсказывать функцию гена, основываясь только на его последовательности.
Прогнозирование гена — один из ключевых этапов аннотации генома , следующий за сборкой последовательности , фильтрацией некодирующих областей и маскированием повторов. [3]
Прогнозирование генов тесно связано с так называемой «проблемой поиска цели», изучающей, как ДНК-связывающие белки ( факторы транскрипции ) находят специфические сайты связывания в геноме . [4] [5] Многие аспекты предсказания структуры генов основаны на современном понимании основных биохимических процессов в клетке , таких как транскрипция генов , трансляция , белок-белковые взаимодействия и процессы регуляции , которые являются предметом активных исследований в различных областях омики , таких как транскриптомика , протеомика , метаболомика и, в более общем плане, структурная и функциональная геномика .
Эмпирические методы
[ редактировать ]В эмпирических (сходстве, гомологии или научно обоснованных) системах поиска генов целевой геном ищет последовательности, сходные с внешними данными, в форме известных экспрессируемых меток последовательностей , информационной РНК (мРНК), белковых продуктов и гомологичных или ортологичные последовательности. Имея последовательность мРНК, легко получить уникальную последовательность геномной ДНК, из которой она должна была быть транскрибирована . Учитывая последовательность белка, семейство возможных кодирующих последовательностей ДНК может быть получено путем обратной трансляции генетического кода . После того, как последовательности ДНК-кандидаты определены, становится относительно простой алгоритмической задачей эффективный поиск совпадений в целевом геноме, полных или частичных, точных или неточных. Учитывая последовательность, алгоритмы локального выравнивания, такие как BLAST , FASTA и Smith-Waterman, ищут области сходства между целевой последовательностью и возможными совпадениями-кандидатами. Совпадения могут быть полными или частичными, точными или неточными. Успех этого подхода ограничен содержанием и точностью базы данных последовательностей.
Высокая степень сходства с известной информационной РНК или белковым продуктом является убедительным доказательством того, что участок целевого генома является геном, кодирующим белок. Однако системное применение этого подхода требует обширного секвенирования мРНК и белковых продуктов. Это не только дорого, но и в сложных организмах в любой момент времени экспрессируется только часть всех генов в геноме организма, а это означает, что внешние доказательства существования многих генов труднодоступны в какой-либо отдельной клеточной культуре. Таким образом, для сбора внешних доказательств существования большинства или всех генов в сложном организме требуется изучение многих сотен или тысяч типов клеток , что представляет дополнительные трудности. Например, некоторые человеческие гены могут экспрессироваться только во время развития эмбриона или плода, что может быть трудно изучить по этическим причинам.
Несмотря на эти трудности, были созданы обширные базы данных транскриптов и последовательностей белков для человека, а также для других важных модельных организмов в биологии, таких как мыши и дрожжи. Например, база данных RefSeq содержит транскрипты и последовательности белков многих различных видов, а система Ensembl всесторонне сопоставляет эти данные с геномами человека и некоторых других. Однако вполне вероятно, что эти базы данных неполны и содержат небольшое, но значительное количество ошибочных данных.
Новые высокопроизводительные технологии секвенирования транскриптома , такие как RNA-Seq и ChIP-секвенирование, открывают возможности для включения дополнительных внешних данных в прогнозирование и проверку генов, а также обеспечивают структурно богатую и более точную альтернативу предыдущим методам измерения экспрессии генов, таким как метка экспрессируемой последовательности или ДНК-микрочип .
Основные проблемы, связанные с предсказанием генов, связаны с ошибками секвенирования необработанных данных ДНК, зависимостью от качества сборки последовательности , обработкой коротких чтений, мутациями сдвига рамки считывания , перекрывающимися генами и неполными генами.
генов важно учитывать горизонтальный перенос генов У прокариот при поиске гомологии последовательностей . Дополнительным важным фактором, недостаточно используемым в современных инструментах обнаружения генов, является существование кластеров генов — оперонов (которые представляют собой функциональные единицы ДНК , содержащие кластер генов под контролем одного промотора ) как у прокариот, так и у эукариот. Большинство популярных детекторов генов рассматривают каждый ген изолированно, независимо от других, что не является биологически точным.
с самого начала Методы
[ редактировать ]Прогнозирование генов Ab Initio — это внутренний метод, основанный на содержании генов и обнаружении сигналов. Из-за неизбежных затрат и трудностей в получении внешних доказательств для многих генов также необходимо прибегнуть к поиску генов ab initio , при котором только последовательность геномной ДНК систематически ищет определенные контрольные признаки генов, кодирующих белок. Эти признаки можно в общих чертах разделить на либо сигналы (специфические последовательности, которые указывают на присутствие поблизости гена), либо содержание (статистические свойства самой последовательности, кодирующей белок). Обнаружение генов ab initio можно было бы более точно охарактеризовать как предсказание генов , поскольку для окончательного установления того, что предполагаемый ген функционален, обычно требуются внешние доказательства.
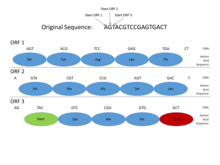
В геномах прокариот гены имеют специфические и относительно хорошо изученные промоторные последовательности (сигналы), такие как бокс Прибнова и транскрипционных факторов сайты связывания , которые легко систематически идентифицировать. Кроме того, последовательность, кодирующая белок, представляет собой одну непрерывную открытую рамку считывания (ORF), длина которой обычно составляет многие сотни или тысячи пар оснований . Статистика стоп-кодонов такова, что даже обнаружение открытой рамки считывания такой длины является достаточно информативным признаком. (Поскольку 3 из 64 возможных кодонов в генетическом коде являются стоп-кодонами, можно было бы ожидать, что стоп-кодон будет появляться примерно через каждые 20–25 кодонов, или 60–75 пар оснований, в случайной последовательности .) Более того, ДНК, кодирующая белок, имеет определенные периодичности и другие статистические свойства, которые легко обнаружить в последовательности такой длины. Эти характеристики делают поиск генов прокариот относительно простым, а хорошо спроектированные системы способны достичь высокого уровня точности.
Обнаружение генов ab initio у эукариот , особенно у таких сложных организмов, как человек, значительно сложнее по нескольким причинам. Во-первых, промотор и другие регуляторные сигналы в этих геномах более сложны и менее понятны, чем у прокариот, что затрудняет их надежное распознавание. Двумя классическими примерами сигналов, идентифицированных с помощью средств поиска эукариотических генов, являются CpG-островки и сайты связывания поли (А)-хвоста .
Во-вторых, механизмы сплайсинга , используемые эукариотическими клетками, означают, что определенная кодирующая белок последовательность в геноме делится на несколько частей ( экзонов ), разделенных некодирующими последовательностями ( интронами ). (Сайты сплайсинга сами по себе являются еще одним сигналом, который часто предназначены для идентификации эукариотических генов.) Типичный ген, кодирующий белок у человека, может быть разделен на дюжину экзонов, каждый из которых имеет длину менее двухсот пар оснований, а некоторые даже короче двадцати. до тридцати. Поэтому гораздо труднее обнаружить периодичности и другие известные свойства содержания белок-кодирующей ДНК у эукариот.
Усовершенствованные специалисты по поиску генов как для прокариотических, так и для эукариотических геномов обычно используют сложные вероятностные модели , такие как скрытые модели Маркова (HMM), для объединения информации из множества различных измерений сигналов и содержания. Система GLIMMER — это широко используемый и высокоточный инструмент для поиска генов прокариот. GeneMark — еще один популярный подход. Для сравнения: эукариотические исследователи генов ab initio добились лишь ограниченного успеха; Яркими примерами являются программы GENSCAN и Geneid . Генеоискатели GeneMark-ES и SNAP основаны на GHMM, как и GENSCAN. Они пытаются решить проблемы, связанные с использованием устройства для поиска генов в последовательности генома, против которой он не был обучен. [7] [8] Несколько недавних подходов, таких как mSplicer, [9] КОНТРАСТ, [10] или mGene [11] также используйте машинного обучения методы , такие как машины опорных векторов, для успешного предсказания генов. Они строят дискриминационную модель, используя скрытые машины опорных векторов Маркова или условные случайные поля, чтобы изучить точную оценочную функцию предсказания генов.
Методы Ab Initio были протестированы, чувствительность некоторых из них приближается к 100%. [3] однако по мере увеличения чувствительности точность снижается из-за увеличения количества ложных срабатываний .
Другие сигналы
[ редактировать ]Среди производных сигналов, используемых для прогнозирования, есть статистика, полученная на основе статистики подпоследовательностей, такая как статистика k-меров , изохора (генетика) или состав/равномерность/энтропия композиционного домена GC, длина последовательности и кадра, интрон/экзон/донор/акцептор/промотор. , сайтов связывания рибосом словарь фрактальная размерность , преобразование Фурье псевдоцифровой ДНК, Z-кривой и некоторые особенности анализа. параметры [12]
Было высказано предположение, что сигналы, отличные от тех, которые непосредственно обнаруживаются в последовательностях, могут улучшить предсказание генов. Например, о роли вторичной структуры в идентификации регуляторных мотивов. сообщалось [13] Кроме того, было высказано предположение, что предсказание вторичной структуры РНК помогает предсказать сайт сплайсинга. [14] [15] [16] [17]
Нейронные сети
[ редактировать ]Искусственные нейронные сети — это вычислительные модели, которые превосходно справляются с машинным обучением и распознаванием образов . Нейронные сети должны быть обучены на примерах данных, прежде чем они смогут обобщать экспериментальные данные и тестироваться на основе эталонных данных. Нейронные сети способны находить приблизительные решения проблем, которые трудно решить алгоритмически, при условии достаточного количества обучающих данных. Применительно к предсказанию генов нейронные сети можно использовать наряду с другими методами ab initio для прогнозирования или идентификации биологических особенностей, таких как сайты сплайсинга. [18] Один подход [19] предполагает использование скользящего окна, которое пересекает данные последовательности с перекрытием. Выходные данные в каждой позиции представляют собой оценку, основанную на том, считает ли сеть, что окно содержит донорный сайт сплайсинга или акцепторный сайт сплайсинга. Окна большего размера обеспечивают большую точность, но также требуют большей вычислительной мощности. Нейронная сеть является примером датчика сигнала, поскольку ее цель — идентифицировать функциональный участок в геноме.
Комбинированные подходы
[ редактировать ]Такие программы, как Maker, сочетают в себе внешние подходы и подходы ab initio, сопоставляя данные о белках и EST с геномом для проверки предсказаний ab initio . Augustus, который можно использовать как часть конвейера Maker, также может включать подсказки в виде выравниваний EST или профилей белков для повышения точности предсказания генов.
Подходы сравнительной геномики
[ редактировать ]Поскольку полные геномы многих различных видов секвенированы, многообещающим направлением в текущих исследованиях по поиску генов является подход сравнительной геномики .
Это основано на том принципе, что силы естественного отбора заставляют гены и другие функциональные элементы подвергаться мутациям с более медленной скоростью, чем остальная часть генома, поскольку мутации в функциональных элементах с большей вероятностью окажут негативное влияние на организм, чем мутации в других местах. Таким образом, гены можно обнаружить путем сравнения геномов родственных видов, чтобы обнаружить это эволюционное давление, направленное на сохранение. Этот подход был впервые применен к геномам мыши и человека с использованием таких программ, как SLAM, SGP и TWINSCAN/N-SCAN и CONTRAST. [20]
Несколько информаторов
[ редактировать ]TWINSCAN исследовал только синтению человека и мыши в поисках ортологичных генов. Такие программы, как N-SCAN и CONTRAST, позволяли включать сопоставления нескольких организмов или, в случае N-SCAN, одного альтернативного организма из цели. Использование нескольких информаторов может привести к значительному повышению точности. [20]
КОНТРАСТ состоит из двух элементов. Первый представляет собой классификатор меньшего размера, идентифицирующий донорные сайты сплайсинга и акцепторные сайты сплайсинга, а также стартовые и стоп-кодоны. Второй элемент предполагает построение полной модели с использованием машинного обучения. Разбиение проблемы на две означает, что для обучения классификаторов можно использовать меньшие целевые наборы данных.и этот классификатор может работать независимо и обучаться с меньшими окнами. Полная модель может использовать независимый классификатор, и вам не придется тратить вычислительное время или усложнять модель на повторную классификацию границ интрон-экзон. В статье, в которой представлен КОНТРАСТ, предлагается классифицировать их метод (а также методы TWINSCAN и т. д.) как сборку генов de novo с использованием альтернативных геномов и отличать ее от ab initio , который использует целевые геномы-информаторы. [20]
Сравнительный поиск генов также можно использовать для переноса высококачественных аннотаций из одного генома в другой. Яркие примеры включают Projector, GeneWise, GeneMapper и GeMoMa. Такие методы теперь играют центральную роль в аннотации всех геномов.
Предсказание псевдогена
[ редактировать ]Псевдогены являются близкими родственниками генов, имеют очень высокую гомологию последовательностей, но не могут кодировать один и тот же белковый продукт. Хотя когда-то их считали побочными продуктами секвенирования генов , но по мере того, как раскрывается их регуляторная роль, они все чаще становятся прогностическими мишенями сами по себе. [21] Прогнозирование псевдогенов использует существующие методы сходства последовательностей и методы ab initio, добавляя при этом дополнительную фильтрацию и методы идентификации характеристик псевдогенов.
Методы сходства последовательностей можно настроить для прогнозирования псевдогенов с использованием дополнительной фильтрации для поиска псевдогенов-кандидатов. Для этого можно использовать обнаружение отключений, которое ищет бессмысленные мутации или мутации сдвига кадра, которые могли бы усечь или свернуть последовательность кодирования, которая в противном случае была бы функциональной. [22] Кроме того, трансляция ДНК в последовательности белков может быть более эффективной, чем просто прямая гомология ДНК. [21]
Датчики содержимого можно фильтровать в соответствии с различиями в статистических свойствах между псевдогенами и генами, такими как уменьшенное количество CpG-островков в псевдогенах или различия в содержании GC между псевдогенами и их соседями. Датчики сигналов также можно настроить на псевдогены, проверяя отсутствие интронов или полиадениновых хвостов. [23]
Метагеномное предсказание генов
[ редактировать ]Метагеномика — это исследование генетического материала, извлеченного из окружающей среды, в результате чего получается информация о последовательностях из пула организмов. Прогнозирование генов полезно для сравнительной метагеномики .
Инструменты метагеномики также попадают в основные категории использования подходов сходства последовательностей (MEGAN4) и методов ab initio (GLIMMER-MG).
Глиммер-МГ [24] является расширением GLIMMER , которое в основном основано на ab initio подходе к поиску генов и использовании обучающих наборов родственных организмов. Стратегия прогнозирования дополняется классификацией и кластеризацией наборов данных о генах перед применением методов прогнозирования генов ab initio. Данные сгруппированы по видам. Этот метод классификации использует методы метагеномной филогенетической классификации. Примером программного обеспечения для этой цели является Phymm, который использует интерполированные марковские модели, и PhymmBL, который интегрирует BLAST в процедуры классификации.
МЕГАН4 [25] использует подход сходства последовательностей, используя локальное выравнивание по базам данных известных последовательностей, но также пытается классифицировать, используя дополнительную информацию о функциональных ролях, биологических путях и ферментах. Как и при прогнозировании генов одного организма, подходы на основе сходства последовательностей ограничены размером базы данных.
FragGeneScan и MetaGeneAnnotator — популярные программы прогнозирования генов, основанные на скрытой модели Маркова . Эти предикторы учитывают ошибки секвенирования, частичные гены и работают для коротких чтений.
Еще один быстрый и точный инструмент для прогнозирования генов в метагеномах — MetaGeneMark. [26] Этот инструмент используется Объединенным институтом генома Министерства энергетики США для аннотирования IMG/M, крупнейшей на сегодняшний день коллекции метагеномов.
См. также
[ редактировать ]- Список программного обеспечения для прогнозирования генов
- Филогенетический след
- Прогнозирование функции белка
- Прогнозирование структуры белка
- Прогнозирование межбелкового взаимодействия
- Псевдоген (база данных)
- Последовательный майнинг
- Сходство последовательностей (гомология)
Ссылки
[ редактировать ]- ^ Слейтор Р.Д. (август 2010 г.). «Обзор текущего состояния стратегий прогнозирования генов эукариот». Джин . 461 (1–2): 1–4. дои : 10.1016/j.gene.2010.04.008 . ПМИД 20430068 .
- ^ Эджигу, Гирум Фитихамлак; Юнг, Джехи (18 сентября 2020 г.). «Обзор компьютерной геномной аннотации последовательностей, полученных с помощью секвенирования следующего поколения» . Биология . 9 (9): 295. doi : 10.3390/biology9090295 . ISSN 2079-7737 . ПМЦ 7565776 . ПМИД 32962098 .
- ^ Jump up to: Перейти обратно: а б Янделл М., Энце Д. (апрель 2012 г.). «Руководство для начинающих по аннотации генома эукариот». Обзоры природы. Генетика . 13 (5): 329–42. дои : 10.1038/nrg3174 . ПМИД 22510764 . S2CID 3352427 .
- ^ Реддинг С., Грин ЕС (май 2013 г.). «Как белки находят определенные цели в ДНК?» . Письма по химической физике . 570 : 1–11. Бибкод : 2013CPL...570....1R . дои : 10.1016/j.cplett.2013.03.035 . ПМЦ 3810971 . ПМИД 24187380 .
- ^ Соколов И.М., Мецлер Р., Пант К., Уильямс М.С. (август 2005 г.). «Целевой поиск N скользящих белков по ДНК» . Биофизический журнал . 89 (2): 895–902. Бибкод : 2005BpJ....89..895S . дои : 10.1529/biophysj.104.057612 . ПМЦ 1366639 . ПМИД 15908574 .
- ^ Мэдиган М.Т., Мартинко Дж.М., Бендер К.С., Бакли Д.Х., Шталь Д. (2015). Брок Биология микроорганизмов (14-е изд.). Бостон: Пирсон. ISBN 9780321897398 .
- ^ «ГенМарк-ЭС» .
- ^ Корф I (май 2004 г.). «Обнаружение генов в новых геномах» . БМК Биоинформатика . 5:59 . дои : 10.1186/1471-2105-5-59 . ПМК 421630 . ПМИД 15144565 .
- ^ Ретч Г., Зонненбург С., Шринивасан Дж., Витте Х., Мюллер К.Р. , Зоммер Р.Дж., Шёлкопф Б. (февраль 2007 г.). «Улучшение аннотации генома Caenorhabditis elegans с помощью машинного обучения» . PLOS Вычислительная биология . 3 (2): е20. Бибкод : 2007PLSCB...3...20R . дои : 10.1371/journal.pcbi.0030020 . ПМК 1808025 . ПМИД 17319737 .
- ^ Гросс СС, До CB, Сирота М, Бацоглу С (20 декабря 2007 г.). «КОНТРАСТ: дискриминационный, свободный от филогении подход к предсказанию генов de novo с использованием множественных информаторов» . Геномная биология . 8 (12): 269 рэндов. дои : 10.1186/gb-2007-8-12-r269 . ПМК 2246271 . ПМИД 18096039 .
- ^ Швайкерт Г., Бер Дж., Зиен А., Целлер Г., Онг К.С., Зонненбург С., Ретч Г. (июль 2009 г.). «mGene.web: веб-сервис для точного компьютерного поиска генов» . Исследования нуклеиновых кислот . 37 (проблема с веб-сервером): W312–6. дои : 10.1093/nar/gkp479 . ПМК 2703990 . ПМИД 19494180 .
- ^ Саис Ю., Рузе П., Ван де Пер Ю. (февраль 2007 г.). «В поисках маленьких: улучшенное предсказание коротких экзонов у позвоночных, растений, грибов и простейших» . Биоинформатика . 23 (4): 414–20. doi : 10.1093/биоинформатика/btl639 . ПМИД 17204465 .
- ^ Хиллер М., Пудимат Р., Буш А., Бакофен Р. (2006). «Использование вторичных структур РНК для поиска мотивов последовательности в направлении одноцепочечных областей» . Исследования нуклеиновых кислот . 34 (17): е117. дои : 10.1093/нар/gkl544 . ПМЦ 1903381 . ПМИД 16987907 .
- ^ Паттерсон DJ, Ясухара К., Руццо В.Л. (2002). «Предсказание вторичной структуры пре-мРНК помогает прогнозировать сайт сплайсинга». Тихоокеанский симпозиум по биокомпьютингу. Тихоокеанский симпозиум по биокомпьютингу : 223–34. ПМИД 11928478 .
- ^ Мараши С.А., Гударзи Х., Садеги М., Эслахчи С., Пезешк Х. (февраль 2006 г.). «Важность информации о вторичной структуре РНК для предсказания донорных и акцепторных сайтов сплайсинга дрожжей с помощью нейронных сетей». Вычислительная биология и химия . 30 (1): 50–7. doi : 10.1016/j.compbiolchem.2005.10.009 . ПМИД 16386465 .
- ^ Мараши С.А., Эслахчи С., Пезешк Х., Садеги М. (июнь 2006 г.). «Влияние структуры РНК на предсказание донорных и акцепторных сайтов сплайсинга» . БМК Биоинформатика . 7 : 297. дои : 10.1186/1471-2105-7-297 . ПМЦ 1526458 . ПМИД 16772025 .
- ^ Рогич, С. (2006). Роль вторичной структуры пре-мРНК в сплайсинге генов у Saccharomyces cerevisiae (PDF) (кандидатская диссертация). Университет Британской Колумбии. Архивировано из оригинала (PDF) 30 мая 2009 г. Проверено 1 апреля 2007 г.
- ^ Гоэл Н., Сингх С., Асери Т.К. (июль 2013 г.). «Сравнительный анализ методов мягких вычислений для предсказания генов». Аналитическая биохимия . 438 (1): 14–21. дои : 10.1016/j.ab.2013.03.015 . ПМИД 23529114 .
- ^ Йохансен, ∅Истейн; Райен, Том; Эфтесоль, Трюгве; Кьосмоен, Томас; Руофф, Питер (2009). «Прогнозирование места сращивания с использованием искусственных нейронных сетей». Методы вычислительного интеллекта для биоинформатики и биостатистики . Lec Not Comp Sci. Том. 5488. стр. 102–113. дои : 10.1007/978-3-642-02504-4_9 . ISBN 978-3-642-02503-7 .
- ^ Jump up to: Перейти обратно: а б с Гросс СС, До CB, Сирота М, Бацоглу С (2007). «КОНТРАСТ: дискриминационный, свободный от филогении подход к предсказанию генов de novo с использованием множественных информаторов» . Геномная биология . 8 (12): 269 рэндов. дои : 10.1186/gb-2007-8-12-r269 . ПМК 2246271 . ПМИД 18096039 .
- ^ Jump up to: Перейти обратно: а б Александр Р.П., Фанг Г., Розовский Дж., Снайдер М., Герштейн М.Б. (август 2010 г.). «Аннотирование некодирующих участков генома». Обзоры природы. Генетика . 11 (8): 559–71. дои : 10.1038/nrg2814 . ПМИД 20628352 . S2CID 6617359 .
- ^ Свенссон О., Арвестад Л., Лагергрен Дж. (май 2006 г.). «Полногеномный обзор биологически функциональных псевдогенов» . PLOS Вычислительная биология . 2 (5): е46. Бибкод : 2006PLSCB...2...46S . дои : 10.1371/journal.pcbi.0020046 . ПМК 1456316 . ПМИД 16680195 .
- ^ Чжан З, Герштейн М (август 2004 г.). «Масштабный анализ псевдогенов в геноме человека». Текущее мнение в области генетики и развития . 14 (4): 328–35. дои : 10.1016/j.где.2004.06.003 . ПМИД 15261647 .
- ^ Келли Д.Р., Лю Б., Делчер А.Л., Поп М., Зальцберг С.Л. (январь 2012 г.). «Прогнозирование генов с помощью Glimmer для метагеномных последовательностей, дополненное классификацией и кластеризацией» . Исследования нуклеиновых кислот . 40 (1): e9. дои : 10.1093/nar/gkr1067 . ПМЦ 3245904 . ПМИД 22102569 .
- ^ Хьюсон Д.Х., Митра С., Рушевей Х.Дж., Вебер Н., Шустер С.К. (сентябрь 2011 г.). «Интегративный анализ последовательностей окружающей среды с использованием MEGAN4» . Геномные исследования . 21 (9): 1552–60. дои : 10.1101/гр.120618.111 . ПМК 3166839 . ПМИД 21690186 .
- ^ Чжу В., Ломсадзе А., Бородовский М. (июль 2010 г.). «Идентификация генов Ab initio в метагеномных последовательностях» . Исследования нуклеиновых кислот . 38 (12): е132. дои : 10.1093/nar/gkq275 . ПМЦ 2896542 . ПМИД 20403810 .
Внешние ссылки
[ редактировать ]- Август
- ФГЕНЕШ. Архивировано 4 января 2013 г. на archive.today.
- GeMoMa — предсказание генов на основе гомологии на основе сохранения положения аминокислот и интронов, а также данных RNA-Seq.
- родился , SGP2
- Glimmer. Архивировано 26 августа 2011 г. в Wayback Machine . GlimmerHMM. Архивировано 18 августа 2011 г. в Wayback Machine.
- ГеномThreader
- Химгеном
- ДжинМарк
- вещь
- мГин
- StarORF — мультиплатформенный веб-инструмент для прогнозирования ORF и получения обратной последовательности комплемента.
- Maker — портативный и легко настраиваемый конвейер аннотаций генома.