Hi-C (метод геномного анализа)

Hi-C — это высокопроизводительный геномный и эпигеномный метод захвата конформации хроматина (3C) . [1] В целом, Hi-C рассматривается как производное от ряда технологий захвата конформации хромосом , включая, помимо прочего, 3C (захват конформации хромосомы), 4C (захват конформации хромосомы на чипе/захват конформации кольцевой хромосомы) и 5C. (захват конформации хромосомы под копией). [1] [2] [3] [4] Hi-C всесторонне обнаруживает полногеномные взаимодействия хроматина в ядре клетки путем сочетания подходов 3C и секвенирования следующего поколения (NGS) и рассматривается как качественный скачок в развитии C-технологии (технологии захвата конформации хромосом) и начало 3D-геномики. [2] [3] [4]
Подобно классическому методу 3C, Hi-C измеряет частоту (в среднем по популяции клеток), с которой два фрагмента ДНК физически связываются в трехмерном пространстве, связывая хромосомную структуру непосредственно с геномной последовательностью. [4] Общая процедура Hi-C включает в себя первое сшивание хроматинового материала с использованием формальдегида . [3] [4] Затем хроматин растворяется и фрагментируется, а взаимодействующие локусы вместе повторно лигируются , чтобы создать геномную библиотеку химерных молекул ДНК . [4] Относительное количество этих химер или продуктов лигирования коррелирует с вероятностью того, что соответствующие фрагменты хроматина взаимодействуют в трехмерном пространстве в популяции клеток. [4] В то время как 3C фокусируется на анализе набора заранее определенных геномных локусов, чтобы предложить исследования конформации представляющих интерес областей хромосомы «один против некоторых», Hi-C обеспечивает профилирование взаимодействия «все против всех», маркируя все фрагментированные фрагменты. хроматин с биотинилированным нуклеотидом перед лигированием. [3] [4] В результате лигированные соединения, отмеченные биотином, могут быть более эффективно очищены с помощью магнитных шариков, покрытых стрептавидином , а данные о взаимодействии хроматина могут быть получены путем прямого секвенирования библиотеки Hi-C. [3] [4]
Анализ данных Hi-C не только раскрывает общую геномную структуру хромосом млекопитающих , но также дает представление о биофизических свойствах хроматина, а также о более специфических, дальних контактах между удаленными геномными элементами (например, между генами и регуляторными элементами ). [4] [5] [6] включая то, как они меняются со временем в ответ на раздражители. [7] В последние годы Hi-C нашел свое применение в самых разных областях биологии, включая рост и деление клеток , регуляцию транскрипции , определение судьбы , развитие, аутоиммунные заболевания и эволюцию генома . [7] [5] [6] Объединив данные Hi-C с другими наборами данных, такими как полногеномные карты модификаций хроматина и профили экспрессии генов, также можно определить функциональную роль конформации хроматина в регуляции и стабильности генома. [4]
История
[ редактировать ]На момент своего создания Hi-C представляла собой технологию с низким разрешением и высоким уровнем шума, которая была способна описывать только области взаимодействия хроматина в пределах размера ячейки в 1 миллион пар оснований (МБ). [1] На создание библиотеки Hi-C также потребовалось несколько дней. [4] [8] а сами наборы данных были низкими как по производительности, так и по воспроизводимости. [9] Тем не менее, данные Hi-C позволили по-новому взглянуть на конформацию хроматина, а также на ядерную и геномную архитектуру, и эти перспективы побудили ученых приложить усилия по модификации этого метода за последнее десятилетие.
В период с 2012 по 2015 год в протокол Hi-C было внесено несколько изменений с расщеплением с 4 резцами. [10] или адаптировали более глубокую глубину секвенирования для получения более высокого разрешения. [8] [9] [11] Использование эндонуклеаз рестрикции , которые режет чаще, или нуклеаз DNaseI и микрококков, также значительно повышало разрешающую способность метода. [12] Совсем недавно (2017 г.) Белагзал и др. описал протокол Hi-C 2.0, который позволил достичь разрешения в килобазах (кб). [12] Ключевой адаптацией к базовому протоколу было удаление этапа солюбилизации SDS после расщепления, чтобы сохранить структуру ядра и предотвратить случайное лигирование фрагментированного хроматина путем лигирования внутри интактных ядер, что легло в основу Hi-C in situ. [12] В 2021 году Лафонтен и др. описал Hi-C 3.0, более высокое разрешение которого было достигнуто за счет усиления сшивки формальдегидом с последующим применением дисукцинимидилглутарата (DSG). [13] В то время как формальдегид захватывает амино- и имино- группы как белков, так и ДНК, NHS-эфиры в DSG реагируют с первичными аминами на белках и могут улавливать взаимодействия аминов. [13] Эти обновления базового протокола позволили ученым рассмотреть более подробные конформационные структуры, такие как хромосомный отсек и топологически ассоциированные домены (TAD), а также конформационные особенности с высоким разрешением, такие как петли ДНК. [12] [13]
На сегодняшний день уже появилось множество производных Hi-C, в том числе Hi-C in situ, low Hi-C, SAFE Hi-C и Micro-C, с отличительными особенностями, связанными с различными аспектами стандарта Hi-C. но основной принцип остался прежним.
Традиционный Hi-C
[ редактировать ]Схема классического рабочего процесса Hi-C выглядит следующим образом: клетки сшиваются формальдегидом; хроматин расщепляется ферментом рестрикции, который образует 5'-выступ ; 5'-выступ заполняется биотинилированными основаниями, и полученная ДНК с тупыми концами лигируется. [1] Продукты лигирования с биотином на стыке отбираются для использования стрептавидина и подвергаются дальнейшей обработке для подготовки библиотеки, готовой для последующего секвенирования. [1]
Парные взаимодействия, которые Hi-C может уловить по всему геному, огромны, поэтому важно проанализировать достаточно большой размер выборки, чтобы уловить уникальные взаимодействия, которые можно наблюдать только у меньшинства общей популяции. [4] Чтобы получить сложную библиотеку продуктов лигирования, которая обеспечит высокое разрешение и глубину данных, в качестве входных данных для Hi-C требуется образец из 20–25 миллионов клеток. [3] [4] Первичные образцы человека, которые могут быть доступны только с меньшим количеством клеток, могут быть использованы для приготовления стандартной библиотеки Hi-C, содержащей всего 1–5 миллионов клеток. [4] Однако использование такого небольшого количества ячеек может быть связано с низкой сложностью библиотеки, что приводит к высокому проценту повторяющихся чтений во время подготовки библиотеки. [4]
Стандарт Hi-C предоставляет данные о парных взаимодействиях с разрешением от 1 до 10 МБ, требует высокой глубины секвенирования, а выполнение протокола занимает около 7 дней. [3] [4] [14]
Формальдегидная сшивка
[ редактировать ]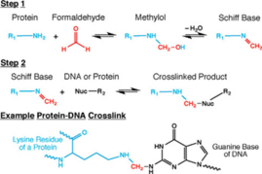
Клеточные и ядерные мембраны обладают высокой проницаемостью для формальдегида. [4] [15] [16] Сшивка формальдегидом часто используется для обнаружения и количественной оценки ДНК-белковых и белок-белковых взаимодействий. [15] Интерес в контексте Hi-C и всех методов, основанных на 3C, представляет способность формальдегида захватывать цис-хромосомные взаимодействия между дистальными сегментами хроматина. [1] [4] [15] [16] Это происходит за счет формирования ковалентных связей между пространственно соседними сегментами хроматина. Формальдегид может реагировать с макромолекулами в два этапа: сначала он реагирует, с нуклеофильной например, группой на основании ДНК, и образует аддукт метилола, который затем преобразуется в основание Шиффа . [15] На втором этапе основание Шиффа, которое может быстро разлагаться, образует метиленовый мостик с другой функциональной группой в другой молекуле. [15] Он также может создать этот метиленовый мостик с помощью небольшой молекулы в растворе, такой как глицин , который в избытке используется для гашения формальдегида в Hi-C. [1] [4] [15] [16] Тушение обычно может оказывать воздействие на формальдегид снаружи клетки. [15] Ключевой особенностью этой двухэтапной реакции сшивания формальдегидом является то, что все реакции обратимы, что жизненно важно для захвата хроматина. [1] [4] [15] [16]
Сшивание является ключевым этапом рабочего процесса захвата хроматина, поскольку функциональным показателем метода является частота, с которой две геномные области сшиваются друг с другом. [4] Таким образом, стандартизация этого этапа важна, и для этого необходимо учитывать потенциальные источники вариаций. [4] Присутствие сыворотки, содержащей высокую концентрацию белка, в культуральной среде может снизить эффективную концентрацию формальдегида, доступного для сшивания хроматина, путем его секвестрации в культуральной среде. [4] Поэтому в тех случаях, когда сыворотка используется в культуре, ее следует удалить для этапа сшивания. [4] Природа клеток, т.е. являются ли они суспензионными или прикрепленными, также является важным фактором для этапа сшивания. [4] Адгезивные клетки связываются с поверхностями с помощью молекулярных механизмов цитоскелета . [4] Было показано, что существует связь между ядерной и клеточной морфологией, поддерживаемой цитоскелетом, которая, если ее изменить, может отрицательно повлиять на глобальную ядерную организацию. [4] Поэтому прикрепившиеся клетки должны быть сшиты, пока они еще прикреплены к поверхности культуры. [4]
Лизис, рестрикционный гидролиз и биотинилирование
[ редактировать ]Клетки лизируют на льду холодным гипотоническим буфером, содержащим хлорид натрия , трис-HCl при pH 8,0 и неионный детергент IGEPAL CA-630 , дополненный ингибиторами протеаз . [4] [16] Ингибиторы протеаз и инкубация на льду помогают сохранить целостность сшитых хроматиновых комплексов эндогенных протеаз. [4] [16] Этап лизиса помогает высвободить нуклеиновый материал из клеток. [1] [4] [16]
После лизиса клеток хроматин солюбилизируется разбавленным SDS, чтобы удалить несшитые белки, открыть хроматин и сделать его более доступным для последующего расщепления, опосредованного эндонуклеазой рестрикции. [4] Если инкубация с SDS превышает рекомендуемые 10 минут, формальдегидные сшивки могут быть обращены вспять, и поэтому за инкубацией с SDS должна немедленно следовать инкубация на льду. [4] Неионогенное моющее средство Triton X-100 используется для гашения SDS, чтобы предотвратить денатурацию фермента на следующем этапе. [4]
Любой фермент рестрикции, который создает 5'-выступ, такой как HindIII, может быть использован для переваривания теперь доступного хроматина в течение ночи. [4] [16] Этот 5'-выступ обеспечивает матрицу, необходимую фрагменту Кленова ДНК -полимеразы I для добавления биотинилированного CTP или АТФ к переваренным концам хроматина. [4] [16] Этот шаг позволяет выбрать продукты лигирования Hi-C для подготовки библиотеки. [4] [16]
Близкое лигирование
[ редактировать ]Лигирование с разбавлением выполняется на фрагментах ДНК, которые все еще сшиты друг с другом, чтобы способствовать внутримолекулярному лигированию фрагментов внутри одного и того же комплекса хроматина вместо событий лигирования между фрагментами в разных комплексах. [4] [16] Поскольку этот этап лигирования происходит между фрагментами ДНК с тупыми концами (поскольку липкие концы заполнены основаниями, меченными биотином), реакции позволяют продолжаться до 4 часов, чтобы компенсировать присущую ей неэффективность. [16] В результате бесконтактного лигирования терминальные сайты HindIII теряются и образуется сайт NheI. [1]
Удаление биотина, разрезание ДНК, выбор размера и восстановление концов.
[ редактировать ]Продукты лигирования, меченные биотином, можно очистить с помощью фенол-хлороформной экстракции ДНК . [4] [16] [17] Для удаления любых фрагментов с меченными биотином концами, которые не были лигированы, используется ДНК-полимераза Т4 с 3'-5'- экзонуклеазной активностью для удаления нуклеотидов с концов таких фрагментов. [4] [16] [18] Этот шаг гарантирует, что ни один из этих нелигированных фрагментов не будет выбран для подготовки библиотеки. [4] [16] Реакцию останавливают ЭДТА и ДНК еще раз очищают с использованием фенол-хлороформной экстракции ДНК. [4] [16]
Идеальный размер фрагментов ДНК для библиотеки секвенирования зависит от используемой платформы секвенирования. [4] [16] ДНК можно сначала разрезать на фрагменты длиной около 300–500 п.н. с помощью обработки ультразвуком . [4] [16] [17] Фрагменты такого размера подходят для высокопроизводительного секвенирования. [4] [16] [17] После обработки ультразвуком фрагменты можно выбрать по размеру с использованием шариков AMPure XP от Beckman Coulter для получения продуктов лигирования с распределением размеров от 150 до 300 пар оснований. [4] [17] Это окно оптимального размера фрагмента для формирования кластера HiSeq. [4] [17]
Сдвиг ДНК вызывает асимметричные разрывы ДНК и должен быть устранен до вытягивания биотина и лигирования адаптера секвенирования. [4] [16] Это достигается за счет использования комбинации ферментов, которые заполняют 5'-выступы и добавляют 5'-фосфатные группы и аденилат к 3'-концам фрагментов, чтобы обеспечить лигирование адаптеров секвенирования. [4] [16]
Сброс биотина
[ редактировать ]Используя избыток стрептавдиновых гранул, таких как раствор стрептавидиновых гранул My-One C1 от Dynabeads , можно извлечь и обогатить биотинилированные продукты лигирования Hi-C. [4] [16] Лигирование парных концевых адаптеров Illumina выполняется во время связывания фрагментов ДНК со стрептавидиновыми шариками. [4] [16] [17] Адсорбция на гранулах повышает эффективность лигирования этих фрагментов ДНК с тупыми концами к адаптерам, поскольку снижает их подвижность. [4] [16] [17]
Подготовка библиотеки и секвенирование
[ редактировать ]После завершения лигирования адаптеров проводят ПЦР- амплификацию библиотеки. [4] [16] Этап ПЦР может привести к появлению большого количества дубликатов в образце продукта лигирования Hi-C низкой сложности в результате чрезмерной амплификации. [4] [16] В результате фиксируется очень мало взаимодействий, и часто это происходит потому, что размер входной выборки имел небольшое количество ячеек. [4] [16] Важно титровать количество циклов, необходимых для получения не менее 50 нг ДНК библиотеки Hi-C для секвенирования. [4] [16] Чем меньше номер цикла, тем лучше, чтобы не было артефактов ПЦР (таких как нецелевые ампликоны, неспецифичность и т. д.). [4] [16] Идеальный диапазон циклов ПЦР составляет 9–15, и более идеально объединить несколько реакций ПЦР, чтобы получить достаточное количество ДНК для секвенирования, чем увеличивать количество циклов для одной реакции ПЦР. [4] [16] Продукты ПЦР снова очищают с использованием гранул AMPure для удаления димеров праймера , а затем определяют количественно перед секвенированием. [4] [16] Области хроматина, которые взаимодействуют друг с другом, затем идентифицируются путем секвенирования парных концов биотинилированных лигированных продуктов. [4] [16]
любая платформа, которая позволяет секвенировать лигированные фрагменты через соединение NheI ( Roche 454) или с помощью парных или парных считываний ( платформы Illumina GA и HiSeq ). Для Hi-C подойдет [4] Перед высокопроизводительным секвенированием качество библиотеки следует проверить с помощью секвенирования по Сэнгеру , при котором считывание длинного секвенирования будет проходить через биотиновое соединение. [4] Тридцать шесть или 50 пар оснований достаточно для идентификации большинства пар взаимодействующих хроматина с использованием парного секвенирования Illumina. [4] Поскольку средний размер фрагментов в библиотеке составляет 250 п.о., парные чтения размером 50 п.о. оказались оптимальными для секвенирования библиотеки Hi-C. [4]
Контроль качества библиотек Hi-C
[ редактировать ]В рабочем процессе подготовки проб Hi-C есть несколько проблемных мест, которые хорошо документированы и описаны. [4] [16] ДНК на различных стадиях можно анализировать на 0,8% агарозном геле, чтобы оценить распределение фрагментов по размерам. [4] [16] Это особенно важно после стрижки на этапах выбора размера. [4] [16] Деградацию ДНК можно отслеживать и по появляющимся в результате мазкам под действием низкомолекулярных продуктов на гелях. [4] [16] Деградация может произойти из-за недостаточного добавления ингибиторов протеазы во время лизиса, активности эндогенной нуклеазы или термической деградации из-за неправильного замораживания. [4] [16] Реакции 3C-ПЦР можно проводить для проверки образования продуктов бесконтактного лигирования. [4] [16]
Варианты
[ редактировать ]Стандарт Hi-C имеет высокую стоимость входного количества клеток, требует глубокого секвенирования, генерирует данные с низким разрешением и страдает от образования избыточных молекул, которые способствуют созданию библиотек низкой сложности, когда количество клеток низкое. [4] [16] [17] Чтобы бороться с этими проблемами и иметь возможность применять этот метод в контекстах, где количество клеток является ограничивающим фактором, например, при работе с первичными клетками человека, с момента первой концептуализации Hi-C было разработано несколько вариантов Hi-C. [3]
Четыре основных класса, под которые подпадают варианты Hi-C: лигирование в разведении, лигирование in situ, одноклеточные системы и системы улучшения с низким уровнем шума. [3] Стандартный Hi-C представляет собой тип лигирования с разбавлением, а другие лигирования с разбавлением включают DNase Hi-C и Capture Hi-C. [3] В отличие от стандарта и Capture Hi-C, для DNase Hi-C требуется всего 2–5 миллионов клеток в качестве входных данных, используется DNaseI для фрагментации хроматина и применяется бесконтактное лигирование с разбавлением в геле. [3] [19] [20] Было показано, что использование DNaseI значительно повышает эффективность и разрешение Hi-C. [3] [19] Capture Hi-C — это метод полногеномного анализа, позволяющий изучить взаимодействия хроматина определенных локусов с использованием гибридизации . захвата целевых геномных областей на основе [20] Впервые он был разработан Мифсудом и др. для картирования дальних контактов промоторов в клетках человека путем создания библиотеки биотинилированных РНК-приманок, нацеленной на 21 841 область промотора. [20] Эти варианты, в дополнение к другим (описанным ниже), представляют собой модификации базовой технологии стандарта Hi-C и адресации и смягчают одно или несколько ограничений исходного метода.
на месте Hi-C
[ редактировать ]Hi-C in situ сочетает в себе стандартный Hi-C с анализом ядерного лигирования, т. е. бесконтактным лигированием, выполняемым в интактных ядрах. [14] [21] Протокол похож на стандартный Hi-C с точки зрения базовой схемы рабочего процесса, но отличается в других отношениях. [14] In situ Hi-C требует от 2 до 5 миллионов клеток по сравнению с идеальными 20–25 миллионами, необходимыми для стандартного Hi-C, и для завершения протокола требуется всего 3 дня по сравнению с 7 днями для стандартного Hi-C. [14] Кроме того, бесконтактное лигирование не происходит в растворе, как в стандартном Hi-C, что снижает частоту случайных, биологически нерелевантных контактов и лигаций, на что указывает более низкая частота контактов митохондриальной и ядерной ДНК в захваченной биотинилированной ДНК. [14] Это достигается за счет того, что ядра остаются нетронутыми на этапе лигирования. [14] Перед лигированием клетки по-прежнему лизируют буфером, содержащим трис-HCl при pH 8,0, хлорид натрия и детергент IGEPAL CA630, но вместо гомогенизации клеточного лизата ядра клеток осаждаются после первоначального лизиса для разрушения клеточной мембраны. [14] После завершения бесконтактного лигирования ядра клеток инкубируют в течение как минимум 1,5 часов при 68 градусах Цельсия для обеспечения проницаемости ядерной мембраны и высвобождения ее ядерного содержимого. [14]
Разрешение, которого можно достичь с помощью Hi-C in situ, может составлять от 950 до 1000 п.о. по сравнению с разрешением от 1 до 10 МБ стандартного Hi-C и разрешением 100 кБ DNase Hi-C. [3] [4] [14] [19] В то время как стандартный Hi-C использует резак из 6 пар оснований, такой как HindIII, для этапа рестрикционного расщепления, Hi-C in situ использует резак из 4 пар оснований, такой как MboI или его изошизомер DpnII (который не чувствителен к метилированию CpG ) для повысить эффективность и разрешающую способность (поскольку в геноме чаще встречаются сайты рестрикции MboI и DpnII). [3] [4] [14] Данные между повторами для Hi-C in situ последовательны и высоко воспроизводимы, с очень меньшим фоновым шумом и демонстрируют четкие взаимодействия хроматина. [3] [14] Однако возможно, что некоторые из уловленных взаимодействий могут быть неточными межмолекулярными взаимодействиями, поскольку ядро плотно упаковано белком и ДНК, поэтому выполнение бесконтактных лигаций в интактных ядрах может устранить мешающие взаимодействия, которые могут формироваться только из-за природы ядерной упаковки и не столько уникальные хромосомные взаимодействия с клеточным функциональным воздействием. [3] [14] Для достижения разрешения данных, описанного Rao et al., также требуется чрезвычайно высокая глубина секвенирования — около 5 миллиардов парных считываний на образец. [3] [14] [22] Существует несколько методов, которые адаптировали концепцию Hi-C in situ, в том числе Sis Hi-C, OCEAN-C и Hi-C захвата in situ. [3] Ниже описаны два наиболее известных метода, основанных на Hi-C. [3]
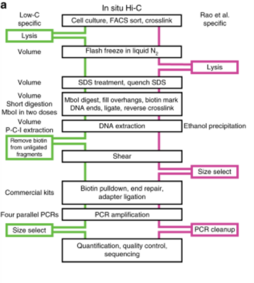
1. Низкий уровень углерода
[ редактировать ]Low-C — это протокол Hi-C in situ, адаптированный для использования при небольшом количестве клеток, который особенно полезен в тех случаях, когда количество клеток является ограничивающим фактором, например, в первичной культуре клеток человека. [23] В этом методе используются незначительные изменения, включая используемые объемы и концентрации, а также время и порядок определенных экспериментальных этапов, чтобы обеспечить создание высококачественных библиотек Hi-C из всего лишь 1000 клеток. [23] Несмотря на потенциал получения пригодных для использования данных высокого разрешения с использованием всего лишь 1000 клеток, Diaz et al. по-прежнему рекомендуется использовать как минимум 1–2 миллиона ячеек, если это возможно, или, если нет, минимум 500 тысяч ячеек. [23] Качество библиотеки сначала оценивалось на платформе Illumina MiSeq (чтение на парных концах 2x84 np), и после прохождения критериев контроля качества (включая низкое количество дубликатов ПЦР) библиотеку секвенировали на Illumina NextSeq (2x80 np на парных концах). [23] В целом, этот метод позволяет обойти проблему, требующую ввода большого количества клеток для Hi-C и высокой глубины секвенирования, необходимой для получения данных с высоким разрешением, но может достигать разрешения только до 5 КБ и не всегда может быть воспроизводимым из-за переменной природы. используемых размеров выборки и полученных на ее основе данных. [23]
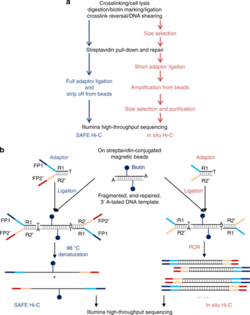
2. БЕЗОПАСНЫЙ Hi-C
[ редактировать ]SAFE Hi-C, или упрощенный, быстрый и экономически эффективный Hi-C, генерирует достаточное количество лигированных фрагментов без амплификации для высокопроизводительного секвенирования. [17] Опубликованные данные In situ Hi-C показывают, что амплификация (на этапе ПЦР для подготовки библиотеки) приводит к смещению амплификации, зависящему от расстояния, что приводит к более высокому соотношению шума к сигналу по сравнению с геномным расстоянием. [17] SAFE Hi-C был успешно использован для создания не требующей амплификации библиотеки лигирования Hi-C in situ всего из 250 тысяч клеток K562 . [17] Фрагменты лигирования имеют длину от 200 до 500 п.н., в среднем около 370 п.н. [17] Все библиотеки продуктов лигирования секвенировали с использованием платформы Illumina HiSeq (2x150 парных концевых считываний). [17] Хотя SAFE Hi-C можно использовать для ввода ячеек всего в 250 тысяч, Niu et al. рекомендуют использовать от 1 до 2 миллионов ячеек. [17] Образцы производят достаточное количество лигатов для секвенирования на четверти дорожки. [17] Было продемонстрировано, что SAFE Hi-C увеличивает сложность библиотеки за счет удаления дубликатов ПЦР, что снижает общий процент уникальных парных чтений. [17] В целом, SAFE Hi-C сохраняет целостность хромосомных взаимодействий, а также снижает необходимость в высокой глубине секвенирования и экономит общие затраты и трудозатраты. [17]
Микро-С
[ редактировать ]Micro-C — это версия Hi-C, которая включает этап расщепления микрококковой нуклеазой (MNase) для изучения взаимодействий между парами нуклеосом , что позволяет разрешать субгеномные структуры TAD в масштабе нуклеосом от 1 до 100. [24] [25] Впервые он был разработан для использования на дрожжах, и было показано, что он сохраняет структурные данные, полученные от стандартного Hi-C, но с большим соотношением сигнал/шум. [24] [25] При использовании эмбриональных стволовых клеток и фибробластов человека на образец было получено от 2,6 до 4,5 миллиардов уникально картированных прочтений. [24] [25] Се и др. проанализировали 2,64 миллиарда считываний эмбриональных стволовых клеток мыши и продемонстрировали увеличение возможностей обнаружения короткодействующих взаимодействий. [24] [25] [26]

Одноэлементный Hi-C
[ редактировать ]Hi-C также был адаптирован для использования с одиночными ячейками, но эти методы требуют высокого уровня знаний для работы и связаны с такими проблемами, как низкое качество данных, покрытие и разрешение. [3]
Анализ данных
[ редактировать ]Химерные продукты лигирования ДНК, генерируемые Hi-C, представляют собой парные взаимодействия хроматина или физические трехмерные контакты внутри ядра. [1] [2] [3] [4] и могут быть проанализированы с помощью различных последующих подходов. Вкратце, данные глубокого секвенирования используются для построения объективных карт взаимодействия хроматина по всему геному. [3] [4] [27] [28] [29] [30] Затем можно использовать несколько различных методов для анализа этих карт с целью выявления структурных закономерностей хромосом и их биологических интерпретаций. Многие из этих подходов к анализу данных также применимы к 3C-секвенированию или другим эквивалентным данным.
Чтение картографии
[ редактировать ]Данные Hi-C, полученные путем глубокого секвенирования, имеют форму традиционного файла FASTQ , и считывания можно сопоставить с интересующим геномом с помощью программного обеспечения для выравнивания последовательностей (например, Bowtie , [31] из [9] [32] и т. д.). [27] [28] Поскольку продукты лигирования Hi-C могут охватывать сотни мегабаз и соединять локусы на разных хромосомах, [3] [4] [27] [28] Выравнивание считывания Hi-C часто является химерным в том смысле, что разные части считывания могут быть выровнены по локусам, удаленным друг от друга, возможно, в разных ориентациях. Длинночитаемые выравниватели (например, minimap2 [33] ) часто поддерживают химерное выравнивание и могут быть непосредственно применены к данным Hi-C с длительным чтением. Выравнивание Hi-C при коротком чтении является более сложной задачей.
Примечательно, что Hi-C создает лигирующие соединения разного размера, но точное положение места лигирования не измеряется. [3] [4] [27] Чтобы обойти эту проблему, итеративное отображение [27] используется, чтобы избежать поиска места соединения, прежде чем появится возможность разделить чтения на два и сопоставить их отдельно для идентификации пар взаимодействия. Идея итеративного картирования состоит в том, чтобы сопоставить как можно более короткую последовательность, чтобы обеспечить уникальную идентификацию пар взаимодействия до достижения места соединения. [27] [28] В результате длинные чтения длиной 25 п.н., начиная с 5'-конца, сначала картируются в геноме, а прочтения, которые не сопоставлены однозначно с одним локусом, расширяются на дополнительные 5 п.н., а затем повторно картируются. [27] Этот процесс повторяется до тех пор, пока все чтения не будут однозначно сопоставлены или пока чтения не будут расширены полностью. [27] [28] Сохраняются только парные концевые чтения, каждая сторона которых уникально сопоставлена с одним геномным локусом. [28] Все остальные парные чтения концов отбрасываются.
Несколько вариантов методов отображения чтения реализованы во многих конвейерах биоинформатики, таких как ICE, [34] HiC-Pro [35] ХИППИ, [36] ПриветCUP, [37] и ТАДбит, [38] для картирования двух частей парного конца, считываемого отдельно, в случае, если эти две части соответствуют различным геномным позициям, тем самым решая проблему, когда считывания охватывают места лигирования. [28]
С увеличенной длиной чтения более поздние конвейеры (например, Juicer [39] и портал данных 4D-нуклеосом [40] ) часто выравнивают короткие чтения Hi-C с помощью алгоритма выравнивания, способного к химерному выравниванию, такого как bwa-mem, [41] хромапа [42] и перетаскивание . Эта процедура вызывает выравнивание один раз и проще, чем итеративное сопоставление.
Назначение и фильтрация фрагментов
[ редактировать ]Каждому картированному прочтению затем присваивается одно местоположение геномного выравнивания в соответствии с его картированным 5'-положением в геноме. [27] Для каждой пары прочтений местоположение назначается только одному из рестрикционных фрагментов , поэтому оно должно находиться в непосредственной близости от сайта рестрикции и на расстоянии, меньшем, чем максимальная длина молекулы. [27] [28] Считывания, картированные на расстоянии, превышающем максимальную длину молекулы от ближайших сайтов рестрикции, являются результатом физического разрушения хроматина или неканонической активности нуклеазы. [27] Поскольку эти чтения также передают информацию о взаимодействиях хроматина, они не отбрасываются, но после назначения геномных местоположений должна быть проведена соответствующая фильтрация, чтобы удалить технический шум в наборе данных. [27] [28] [29] [30]
В зависимости от того, попадает ли пара чтения в один и тот же или в разные фрагменты ограничения, применяются разные критерии фильтрации. Если парные прочтения соответствуют одному и тому же рестрикционному фрагменту, они, скорее всего, представляют собой несвязанные свисающие концы или кольцевые фрагменты, которые неинформативны и поэтому удаляются из набора данных. [27] [28] Эти чтения также могут представлять собой артефакты ПЦР, непереваренные фрагменты хроматина или просто чтения с низким качеством выравнивания. [8] [28] Независимо от происхождения, чтения, сопоставленные с одним и тем же фрагментом, считаются «ложными сигналами». [28] и обычно отбрасываются перед последующей обработкой.
Остальные парные прочтения, сопоставленные с отдельными фрагментами рестрикции, также фильтруются для исключения идентичных/избыточных продуктов ПЦР, и это достигается путем удаления прочтений, имеющих одну и ту же последовательность или положения выравнивания 5'. [27] Дополнительные уровни фильтрации также могут быть применены для достижения экспериментальных целей. Например, потенциальные непереваренные сайты рестрикции можно целенаправленно отфильтровывать, а не пассивно идентифицировать, удаляя прочтения, сопоставленные с одной и той же хромосомной цепью, с небольшим расстоянием (определяемым пользователем, основанным на опыте) между ними. [27]

Биннинг и фильтрация на уровне бинов
[ редактировать ]На основе координат средней точки рестрикционные фрагменты Hi-C объединены в фиксированные геномные интервалы с размерами ячеек от 40 КБ до 1 МБ. [27] Обоснование этого подхода заключается в том, что за счет уменьшения сложности данных и уменьшения количества потенциальных полногеномных взаимодействий на бин, геномные бины позволяют создавать более надежные и менее зашумленные сигналы в форме контактных частот на за счет разрешения (хотя длина ограничительного фрагмента по-прежнему остается предельным физическим пределом разрешения Hi-C). [27] [28] Взаимодействия между бункерами агрегируются путем простого суммирования, хотя с годами были разработаны более целенаправленные и информативные методы для дальнейшего улучшения сигнала. [27] Один из таких методов описан Rao et al. Целью является расширение предела размера бункера до все меньших и меньших бункеров, в конечном итоге имея > 80% бункеров, охваченных 1000 считываний каждый, что значительно повышает разрешающую способность результатов окончательного анализа. [14]
Фильтрация на уровне ячеек, как и фильтрация на уровне фрагментов, также применяется для удаления экспериментальных артефактов из полученных данных. Бины с высоким уровнем шума и низкими сигналами удаляются, поскольку они обычно представляют собой сильно повторяющееся геномное содержимое вокруг теломер и центромер . [27] Это делается путем сравнения сумм отдельных интервалов с суммой всех интервалов и удаления нижнего 1% интервалов или использования дисперсии в качестве меры шума. [27] Ячейки с низким покрытием или ячейки на три стандартных отклонения ниже центра логарифмически нормального распределения (которое соответствует общему количеству контактов на одну геномную ячейку) удаляются с помощью фильтра MAD-max (максимально допустимое медианное абсолютное отклонение). [43] [44] После биннинга данные Hi-C будут сохранены в формате симметричной матрицы. [27] [28] [29] [30]
Совсем недавно было предложено множество подходов для предопределения оптимального размера контейнера для различных экспериментов с Hi-C. Ли и др. в 2018 году описал deDoc, метод, в котором размер ячейки выбирается такой, при котором структурная энтропия матрицы Hi-C достигает стабильного минимума. [45] QuASAR, с другой стороны, предлагает немного более качественную оценку и сравнивает оценки повторов образцов (учитывая, что реплики действительно включены для экспериментальных целей), чтобы найти максимально полезное разрешение. [46] Некоторые публикации [8] [47] также попытались оценить частоты взаимодействия на уровне одного фрагмента, где более высокий охват может быть достигнут даже при меньшем количестве чтений. ПриветCPlus, [48] инструмент, разработанный Чжаном и др. в 2018 году способен вменять матрицы Hi-C, аналогичные исходным, используя только 1/16 исходных чтений. [48]
Балансировка/нормализация
[ редактировать ]Балансировка относится к процессу коррекции смещения полученных данных Hi-C и может быть явным или неявным. [27] [28] Методы явной балансировки требуют явного определения систематических ошибок, которые, как известно, связаны с чтением Hi-C (или любым методом высокопроизводительного секвенирования в целом), включая возможность сопоставления чтения, содержимое GC , а также длину отдельного фрагмента. [27] [28] Поправочный коэффициент сначала вычисляется для каждого из рассматриваемых отклонений, затем для каждой их комбинации, а затем применяется к количеству чтений на геномный бин. [27] [28]
Однако некоторые смещения могут иметь неизвестное происхождение, и в этом случае вместо этого используется подход неявной балансировки. Неявное балансирование основано на предположении, что каждый геномный локус должен иметь «равную видимость», что предполагает, что сигнал взаимодействия в каждом геномном локусе в данных Hi-C должен составлять одну и ту же общую сумму. [28] Один подход, называемый итеративной коррекцией, использует алгоритм балансировки Синкхорна – Кноппа. [49] и пытается сбалансировать симметричную матрицу, используя вышеупомянутое предположение (путем выравнивания суммы каждой строки и столбца в матрице). [27] [28] [49] Алгоритм итеративно чередует два шага: 1) деление каждой строки на ее среднее значение и 2) деление каждого столбца на его среднее значение, которые в конечном итоге гарантированно сходятся и не оставляют явно высоких строк или столбцов в матрице взаимодействия. [27] [49] Существуют также другие вычислительные методы для нормализации систематических ошибок, присущих данным Hi-C, включая нормализацию последовательных компонентов (SCN), [50] подход балансировки матрицы Найта-Руиса, [14] [51] и нормализация разложения собственных векторов (ICE). [34] В конце концов, как явные, так и неявные методы коррекции смещения дают сопоставимые результаты. [27]
Анализ и интерпретация данных
[ редактировать ]С помощью объединенной полногеномной матрицы взаимодействия можно идентифицировать и биологически интерпретировать общие закономерности взаимодействия, наблюдаемые в геномах млекопитающих, в то время как более редкие и менее часто наблюдаемые закономерности, такие как кольцевые хромосомы и кластеризация центромер, могут потребовать дополнительных, специально адаптированных методов для идентификации. .
1. Соотношение цис/транс взаимодействия
[ редактировать ]Цис / транс-взаимодействия являются одним из двух наиболее сильных паттернов взаимодействия, наблюдаемых на картах Hi-C. [27] Они не локус-специфичны и поэтому считаются паттерном уровня генома. [27] Обычно более высокая частота взаимодействия наблюдается в среднем для пар локусов, находящихся на одной хромосоме (в цис), чем для пар локусов, находящихся на разных хромосомах (в транс). [27] В матрицах взаимодействия Hi-C цис/транс-взаимодействия выглядят как квадратные блоки, центрированные по диагонали, одновременно совпадающие с отдельными хромосомами. [27] Поскольку эта закономерность относительно одинакова для разных видов и типов клеток, ее можно использовать для оценки качества данных. Более шумный эксперимент из-за случайного фонового лигирования или любого неизвестного фактора приведет к более низкому соотношению цис- и транс-взаимодействий (поскольку ожидается, что шум будет влиять как на цис-, так и на транс-взаимодействия в одинаковой степени), а эксперименты высокого качества обычно имеют соотношение цис/транс-взаимодействия составляет от 40 до 60 для генома человека. [27]
2. Частота взаимодействия, зависящая от расстояния
[ редактировать ]Этот паттерн относится к зависимому от расстояния затуханию частот взаимодействия на уровне генома и представляет собой второй из двух самых сильных паттернов взаимодействия Hi-C. [27] По мере уменьшения частот взаимодействия между цис-взаимодействующими локусами (в результате дальнейшего расстояния между ними) можно наблюдать постепенное снижение частоты взаимодействия при удалении от диагонали в матрице взаимодействия. [27]
Различные полимерные модели [52] [53] существуют для статистической характеристики свойств пар локусов, разделенных заданным расстоянием, но дискретное объединение и подгонка непрерывных функций являются двумя распространенными способами анализа частот взаимодействия между точками данных, зависящих от расстояния. [27] Во-первых, частоты взаимодействия можно группировать на основе их геномного расстояния, затем к данным подгоняется непрерывная функция с использованием информации о среднем значении каждого интервала. [27] Полученная функция затухания отображается на логарифмическом графике, так что линейную линию можно использовать для представления степенного затухания, предсказанного полимерными моделями. [52] [53] Однако зачастую простой модели полимера оказывается недостаточно для полного представления частот взаимодействия, зависящих от расстояния, и в этот момент могут возникнуть более сложные функции распада, что может повлиять на воспроизводимость данных из-за присутствия локус-специфичных, а не геномных -широкие закономерности, наблюдаемые в матрице Hi-C (которые не учитываются полимерными моделями). [27] [52] [53]
3. Хроматиновые отсеки
[ редактировать ]Самый сильный локус-специфичный паттерн, обнаруженный на картах Hi-C, - это компартменты хроматина , [1] который принимает форму клетчатого или «шахматного» рисунка на матрице взаимодействия с чередующимися блоками размером от 1 до 10 Мб (что позволяет легко извлекать их даже в экспериментах с очень низкой выборкой) в геном человека. [27] [28] [30] Эту закономерность можно обнаружить как на высоких, так и на низких частотах. Поскольку хромосомы состоят из двух типов геномных участков, чередующихся по длине отдельных хромосом, частоты взаимодействия между двумя однотипными участками и частоты взаимодействия между двумя участками разных типов могут существенно различаться. [27] [28]
Определение активного (А) и неактивного (В) компартментов хроматина основано на анализе главных компонентов , впервые установленном Lieberman-Aiden et al. в 2009 году. [1] [27] [28] [30] Их подход рассчитывал корреляцию отношения наблюдаемого и ожидаемого сигналов (полученного из нормализованной по расстоянию контактной матрицы) матрицы Hi-C и использовал знак первого собственного вектора для обозначения положительных и отрицательных частей результирующего графика как A и Отсеки B соответственно. [1] [27] [28] [30] Многие геномные исследования показали, что компартменты хроматина коррелируют с состояниями хроматина, такими как плотность генов , доступность ДНК, содержание GC, время репликации и гистонов . метки [1] [27] [28] [30] Таким образом, компартменты типа A более конкретно определены как представляющие густогенные области эухроматина , тогда как компартменты типа B представляют собой гетерохроматические области с меньшей активностью генов. [27] [28] [30] В целом компартменты хроматина дают представление об общих принципах организации интересующего генома.
За последнее десятилетие было разработано все больше и больше биоинформатических инструментов, способных выполнять вызов отсеков, включая HOMER, [54] ПриветТК Р, [35] и CscoreTool. [55] Хотя каждый из них имеет свои собственные различия и оптимизации, сделанные на основе исходного подхода 2009 года, их базовые протоколы по-прежнему полагаются на анализ главных компонентов.
4. Топологически ассоциированные домены (TAD).
[ редактировать ]TAD представляют собой структуры суб-Mb, которые могут обладать функциями генной регуляции, такими как локальные промотор - энхансер . взаимодействия [27] В более общем смысле, TADs рассматриваются как возникающее свойство лежащих в основе биологических механизмов, которое определяет TAD как экструзии петель, компартментализации или любой динамический геномный паттерн, а не статическую структурную особенность генома. [56] Таким образом, TAD представляют собой регуляторное микроокружение и обычно отображаются на карте Hi-C как блоки областей с высокой степенью самовзаимодействия, в которых частоты взаимодействия внутри региона значительно выше, чем частоты взаимодействия между двумя соседними областями. [27] [28] [30] В матрицах взаимодействия Hi-C TAD представляют собой квадратные блоки повышенных частот взаимодействия, центрированные по диагонали. [27] Однако это всего лишь упрощенное описание, и выявление фактической закономерности требует гораздо большей статистической обработки и оценки.
Один из подходов к выявлению TAD был описан Dixon et al., [9] где они сначала рассчитали (в пределах некоторого геномного диапазона) разницу между средними вышестоящими взаимодействиями и средними нисходящими взаимодействиями каждого интервала в матрице. [9] Эта разница затем была преобразована в статистику хи-квадрат, основанную на скрытой модели Маркова , и любое резкое изменение этого значения хи-квадрат, называемого индексом направленности, будет определять границы TAD. [9] [27] Альтернативно, можно просто взять соотношение между средними взаимодействиями вверх и вниз по течению, чтобы определить границы TAD, как это сделали Наумова и др. [57]
Другой подход заключается в вычислении средних частот взаимодействия по каждому интервалу, опять же в пределах некоторого заранее определенного геномного диапазона. [27] [28] [58] Полученное значение называется показателем изоляции и может рассматриваться как среднее значение квадрата, скользящего по диагонали матрицы (Крейн и др.). [58] Ожидается, что это значение будет ниже на границах TAD; таким образом, можно использовать стандартные статистические методы для поиска локальных минимумов (границ) и определять области между последовательными границами как TAD. [27] [28] [58]
Однако, как сегодня все больше признается, TAD представляют собой иерархическую серию структур, которые невозможно полностью охарактеризовать одномерными оценками, полученными с помощью предыдущих методов. [28] Повышенное разрешение, доступное в новых наборах данных, теперь может напрямую решать проблему TAD с помощью подходов многомасштабного анализа. Как впервые представил Арматус, [59] могут быть идентифицированы домены, специфичные для разрешения, и может быть рассчитан консенсусный набор доменов, сохраняющийся для разных разрешений, [28] [59] что преобразует проблему вызова TAD в оптимизацию скоринговых функций на основе их локальных плотностей взаимодействия. [59] Варианты этого подхода с различными целевыми функциями, такие как «Взрыв лавы», [60] г-нТАДФиндер, [61] 3DNetMod, [62] и Матрешка, [63] также разработаны для повышения производительности вычислений на наборах данных с более высоким разрешением.
5. Взаимодействие точек
[ редактировать ]С биологической точки зрения регуляторные взаимодействия обычно происходят в гораздо меньших масштабах, чем TAD, и два геномных элемента могут активировать/ингибировать экспрессию гена на расстоянии всего лишь 1 т.п.н. [27] Следовательно, точечные взаимодействия важны для интерпретации карт Hi-C и, как ожидается, проявятся как локальное увеличение вероятности контакта. [27] [28] Однако все современные методологии идентификации точечных взаимодействий носят неявный характер, поскольку они не указывают, как должно выглядеть точечное взаимодействие. [27] [28] Вместо этого точечные мутации идентифицируются как выбросы с более высокими частотами взаимодействия, чем ожидалось в матрице Hi-C, учитывая, что фоновая модель состоит только из самых сильных сигналов, таких как функции затухания расстояния. [27] [28] Фоновую модель можно оценить и построить, используя как локальные распределения сигналов, так и глобальные подходы (т. е. общехромосомные/геномные подходы). [28] Многие из вышеупомянутых пакетов биоинформатики включают алгоритмы для определения точечных взаимодействий. Короче говоря, вычисляется значимость отдельного парного взаимодействия, и значительно высокие выбросы корректируются для многократного тестирования, прежде чем они будут признаны действительно информативными точечными взаимодействиями. [27] Полезно дополнить выявленные точечные взаимодействия дополнительными доказательствами, такими как анализ показателей обогащения и биологических повторов, чтобы указать, что эти взаимодействия действительно имеют биологическое значение. [27]
Использование
[ редактировать ]Разработка
[ редактировать ]1. Деление клеток
[ редактировать ]Hi-C может выявить изменения конформации хроматина во время деления клеток. В интерфазе хроматины обычно рыхлые и подвижные, что позволяет осуществлять регуляцию транскрипции и другие регуляторные действия. [64] При вступлении в митоз и деление клетки хроматины компактно сворачиваются в плотные цилиндрические хромосомы. [64] За последние пять лет разработка одноклеточного Hi-C позволила отобразить весь трехмерный структурный ландшафт хроматинов/хромосом на протяжении клеточного цикла , и многие исследования обнаружили, что эти идентифицированные геномные домены остаются неизменными в интерфазе. стираются механизмами молчания, когда клетка вступает в митоз. [65] [66] Когда митотическое деление завершается и клетка снова входит в интерфазу, наблюдается восстановление трехмерных структур хроматина и регуляция транскрипции. [65]
2. Регуляция транскрипции и определение судьбы
[ редактировать ]Предполагалось, что дифференцировка эмбриональных стволовых клеток (ЭСК) и индуцированных плюрипотентных стволовых клеток (ИПСК) в различные линии зрелых клеток сопровождается глобальными изменениями в хромосомных структурах и, следовательно, в динамике взаимодействия, позволяющей регулировать активацию/молчание транскрипции. [3] Для исследования этого исследовательского вопроса можно использовать стандарт Hi-C.
В 2015 году Диксон и др. [11] применили стандарт Hi-C для захвата глобальной трехмерной динамики ЭСК человека во время их дифференцировки в клетки «высокой пятерки» . Благодаря способности Hi-C отображать динамические взаимодействия в TAD, связанных с дифференцировкой, исследователи обнаружили увеличение количества сайтов DHS, способности связывания CTCF , активных модификаций гистонов и экспрессии целевых генов в этих интересующих TAD, а также обнаружили значительные участие основных факторов плюрипотентности, таких как OCT4 , NANOG и SOX2, в сети взаимодействия во время перепрограммирования соматических клеток . [11] С тех пор Hi-C был признан одним из стандартных методов исследования регуляторной активности транскрипции и подтвердил, что архитектура хромосом тесно связана с судьбой клеток. [11] [67]
3. Рост и развитие
[ редактировать ]Соматический рост и развитие млекопитающих начинается с оплодотворения сперматозоида стадия и яйцеклетки , за которым следуют стадия зиготы , 2-клеточная, 4-клеточная и 8-клеточная стадии, стадия бластоцисты и, наконец, эмбриона . [68] Hi-C позволил изучить комплексную архитектуру генома во время роста и развития, поскольку оба sis-Hi-C [69] и на месте Hi-C [70] сообщили, что TAD и геномные компартменты A и B явно не присутствуют и, по-видимому, менее хорошо структурированы в клетках ооцитов. [69] [70] Эти структурные особенности хроматина постепенно устанавливаются от более слабых частот к более чистым и более частым точкам данных после оплодотворения по мере продвижения стадий развития. [69] [70]
Эволюция генома
[ редактировать ]Поскольку в последние годы данные о трехмерных структурах генома становятся все более распространенными, Hi-C начинает использоваться как средство отслеживания эволюционных структурных особенностей/изменений. Геномные однонуклеотидные полиморфизмы (SNP) и TAD обычно консервативны у разных видов. [71] наряду с фактором CTCF в эволюции домена хроматина. [72] Однако с помощью методов Hi-C были выявлены и другие факторы, способствующие структурной эволюции в 3D-архитектуре. К ним относятся сходство частоты использования кодонов (CUFS), [73] совместная регуляция гена паралога, [74] и пространственно развивающиеся ортологичные модули (SCOM). [75] Для крупномасштабной эволюции доменов хромосомные транслокации , синтенные области, а также области геномной перестройки были относительно консервативными. [2] [67] [72] [76] [77] Эти результаты подразумевают, что технологии Hi-C способны обеспечить альтернативную точку зрения на эукариот . древо жизни [3]
Рак
[ редактировать ]В нескольких исследованиях использовалось использование Hi-C для описания и изучения архитектуры хроматина при различных видах рака и их влияния на патогенез заболеваний. Клетген и др. использовали in situ Hi-C для изучения острого лимфобластного лейкоза Т-клеток (T-ALL) и обнаружили событие слияния TAD, которое удалило участок изоляции CTCF, что позволило промотору онкогена MYC напрямую взаимодействовать с дистальным суперэнхансером . [78] Фанг и др. также показали, как происходит специфическое усиление или потеря изоляции хроматина T-ALL, что изменяет силу TAD-архитектуры генома, используя in situ Hi-C. [79] Low-C использовался для картирования структуры хроматина первичных B-клеток пациента с диффузной крупноклеточной B-клеточной лимфомой и использовался для обнаружения высоких структурных различий хромосом между пациентом и здоровыми B-клетками. [23] В целом, применение Hi-C и его вариантов в исследованиях рака дает уникальное понимание молекулярных основ движущих факторов клеточных аномалий. [23] [78] [79] Это может помочь объяснить биологические явления (высокая экспрессия MYC в T-ALL) и помочь в разработке лекарств, нацеленных на механизмы, уникальные для раковых клеток. [23] [78] [79]
Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г час я дж к л м н тот Либерман-Эйден, Э ; ван Беркум, Нидерланды; Уильямс, Л; Имакаев М; Рагочи, Т; Рассказываю, А; Амит, я; Ладжуа, БР; Сабо, Пи Джей; Доршнер, Миссури; Сандстром, Р; Бернштейн, Б ; Бендер, Массачусетс; Грудин, М ; Гнирке, А; Стаматояннопулос, Дж .; Мирный, Луизиана; Ландер, ES ; Деккер, Дж (9 октября 2009 г.). «Комплексное картирование долгосрочных взаимодействий раскрывает принципы свертывания человеческого генома» . Наука . 326 (5950): 289–93. Бибкод : 2009Sci...326..289L . дои : 10.1126/science.1181369 . ПМЦ 2858594 . ПМИД 19815776 .
- ^ Jump up to: а б с д Лин, Да; Хун, Пинг; Чжан, Сихэн; Сюй, Вайзе; Джамал, Мухаммед; Ян, Кеджи; Лей, Иньин; Ли, Лян; Жуань, Ицзюнь; Фу, Чжэнь Ф.; Ли, Голян; Цао, Банда (май 2018 г.). «Hi-C, основанный только на переваривании и лигировании, является эффективным и экономичным методом захвата конформации хромосом» . Природная генетика . 50 (5): 754–763. дои : 10.1038/s41588-018-0111-2 . ISSN 1546-1718 . ПМИД 29700467 . S2CID 13740808 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С Конг, Сиюань; Чжан, Юбо (1 февраля 2019 г.). «Расшифровка Hi-C: от 3D-генома к функции» . Клеточная биология и токсикология . 35 (1): 15–32. дои : 10.1007/s10565-018-09456-2 . ISSN 1573-6822 . ПМИД 30610495 . S2CID 57427743 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо топор является тот нет бб до нашей эры др. быть парень бг чб с минет БК с бм млрд быть б.п. БК бр бс БТ этот бв б бх к Белтон, Джон-Мэтью; МакКорд, Рэйчел Паттон; Гибкус, Йохан; Наумова, Наталья; Чжан, Е; Деккер, Иов (ноябрь 2012 г.). «Hi-C: комплексный метод определения конформации геномов» . Методы . 58 (3): 268–276. дои : 10.1016/j.ymeth.2012.05.001 . ISSN 1046-2023 . ПМЦ 3874846 . ПМИД 22652625 .
- ^ Jump up to: а б Иген, Кайл П. (июнь 2018 г.). «Принципы хромосомной архитектуры, раскрытые Hi-C» . Тенденции биохимических наук . 43 (6): 469–478. дои : 10.1016/j.tibs.2018.03.006 . ISSN 0968-0004 . ПМК 6028237 . ПМИД 29685368 .
- ^ Jump up to: а б Ким, Кюкванг; Ким, Муён; Ким, Юбин; Ли, Донсунг; Юнг, Инкьюнг (1 января 2022 г.). «Hi-C как молекулярный дальномер для изучения геномных перестроек» . Семинары по клеточной биологии и биологии развития . 121 : 161–170. дои : 10.1016/j.semcdb.2021.04.024 . ISSN 1084-9521 . ПМИД 33992531 . S2CID 234746398 .
- ^ Jump up to: а б Беррен, Оливер С.; Рубио Гарсия, Аркадио; Хавьер, Биола-Мария; Радуга, Дэниел Б.; Кэрнс, Джонатан; Купер, Николас Дж.; Ламбурн, Джон Дж.; Шофилд, Эллен; Кастро Допико, Ксакин; Феррейра, Рикардо К.; Коулсон, Ричард; Берден, Фрэнсис; Роулстон, София П.; Даунс, Кейт; Вингетт, Стивен В. (4 сентября 2017 г.). «Хромосомные контакты в активированных Т-клетках идентифицируют гены-кандидаты аутоиммунных заболеваний» . Геномная биология . 18 (1): 165. дои : 10.1186/s13059-017-1285-0 . ISSN 1474-760X . ПМК 5584004 . ПМИД 28870212 .
- ^ Jump up to: а б с д Джин, Фулай; Ли, Ян; Диксон, Джесси Р.; Сельварадж, Сиддарт; Йе, Чжэнь; Ли, А Янг; Йен, Чиа-Ань; Шмитт, Энтони Д.; Эспиноза, Селсо; Рен, Бинг (14 ноября 2013 г.). «Карта трехмерного интерактома хроматина в клетках человека в высоком разрешении» . Природа . 503 (7475): 290–294. Бибкод : 2013Natur.503..290J . дои : 10.1038/nature12644 . ISSN 0028-0836 . ПМЦ 3838900 . ПМИД 24141950 .
- ^ Jump up to: а б с д и ж Диксон-младший; Сельварадж, С; Юэ, Ф; Ким, А; Ли, Ю; Шен, Ю; Ху, М; Лю, Дж.С.; Рен, Б. (11 апреля 2012 г.). «Топологические домены в геномах млекопитающих, выявленные путем анализа взаимодействий хроматина» . Природа . 485 (7398): 376–80. Бибкод : 2012Natur.485..376D . дои : 10.1038/nature11082 . ПМЦ 3356448 . ПМИД 22495300 .
- ^ Секстон, Том; Яффе, Эйтан; Кенигсберг, Ефрем; Бантиньи, Фредерик; Леблан, Бенджамин; Хойхман, Майкл; Парринелло, Хьюз; Танай, Амос; Кавалли, Джакомо (3 февраля 2012 г.). «Принципы трехмерной складки и функциональной организации генома дрозофилы» . Клетка . 148 (3): 458–472. дои : 10.1016/j.cell.2012.01.010 . ISSN 0092-8674 . ПМИД 22265598 . S2CID 17364610 .
- ^ Jump up to: а б с д Диксон, Джесси Р.; Юнг, Инкьюнг; Сельварадж, Сиддарт; Шен, Инь; Антосевич-Бурже, Джессика Э.; Ли, А Янг; Йе, Чжэнь; Ким, Одри; Раджагопал, Ниша; Се, Вэй; Диао, Яруи; Лян, Цзин; Чжао, Хуэйминь; Лобаненков Виктор Викторович; Экер, Джозеф Р.; Томсон, Джеймс А.; Рен, Бинг (февраль 2015 г.). «Реорганизация архитектуры хроматина во время дифференцировки стволовых клеток» . Природа . 518 (7539): 331–336. Бибкод : 2015Natur.518..331D . дои : 10.1038/nature14222 . ISSN 1476-4687 . ПМЦ 4515363 . ПМИД 25693564 .
- ^ Jump up to: а б с д Белагзал, Хауда; Деккер, Иов; Гибкус, Йохан Х. (1 июля 2017 г.). «Hi-C 2.0: оптимизированная процедура Hi-C для полногеномного картирования конформации хромосом» . Методы . 123 : 56–65. дои : 10.1016/j.ymeth.2017.04.004 . ISSN 1046-2023 . ПМЦ 5522765 . ПМИД 28435001 .
- ^ Jump up to: а б с Лафонтен, Денис Л.; Ян, Лиян; Деккер, Иов; Гибкус, Йохан Х. (июль 2021 г.). «Hi-C 3.0: Улучшенный протокол для полногеномного захвата конформации хромосом» . Текущие протоколы . 1 (7): e198. дои : 10.1002/cpz1.198 . ISSN 2691-1299 . ПМК 8362010 . PMID 34286910 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот Рао, Сухас СП; Хантли, Мириам Х.; Дюран, Нева С.; Стаменова Елена К.; Бочков Иван Д.; Робинсон, Джеймс Т.; Сэнборн, Адриан Л.; Махол, Идо; Омер, Арина Д.; Ландер, Эрик С.; Эйден, Эрез Либерман (18 декабря 2014 г.). «Трехмерная карта генома человека с разрешением в килобазах раскрывает принципы образования петель хроматина» . Клетка . 159 (7): 1665–1680. дои : 10.1016/j.cell.2014.11.021 . ISSN 0092-8674 . ПМК 5635824 . ПМИД 25497547 .
- ^ Jump up to: а б с д и ж г час я Хоффман, Элизабет А.; Фрей, Брайан Л.; Смит, Ллойд М.; Обл, Дэвид Т. (30 октября 2015 г.). «Сшивка формальдегидом: инструмент для изучения хроматиновых комплексов» . Журнал биологической химии . 290 (44): 26404–26411. дои : 10.1074/jbc.R115.651679 . ISSN 0021-9258 . ПМЦ 4646298 . ПМИД 26354429 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь ван Беркум, Нинка Л.; Либерман-Айден, Эрез ; Уильямс, Луиза; Имакаев Максим; Гнирке, Андреас; Мирный, Леонид А.; Деккер, Иов; Ландер, Эрик С. (6 мая 2010 г.). «Hi-C: метод изучения трехмерной архитектуры геномов» . Журнал визуализированных экспериментов (39): 1869. doi : 10.3791/1869 . ISSN 1940-087X . ПМК 3149993 . ПМИД 20461051 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р Ню, Лунцзянь; Шен, Вэй; Хуан, Инчжан; Он, На; Чжан, Юэдун; Сунь, Цзялей; Ван, Цзин; Цзян, Даксин; Ян, Манюн; Цзе, Ю Чунг; Ли, Ли; Хоу, Чуньхуэй (19 июля 2019 г.). «При подготовке библиотеки без амплификации с помощью SAFE Hi-C используются продукты лигирования для глубокого секвенирования для улучшения традиционного анализа Hi-C» . Коммуникационная биология . 2 (1): 267. doi : 10.1038/s42003-019-0519-y . ISSN 2399-3642 . ПМК 6642088 . ПМИД 31341966 .
- ^ Детч, П.В.; Чан, Г.Л.; Хазелтин, Вашингтон (10 мая 1985 г.). «Экзонуклеаза ДНК-полимеразы Т4 (3'–5'), фермент для обнаружения и количественного определения стабильных повреждений ДНК: пример ультрафиолетового света» . Исследования нуклеиновых кислот . 13 (9): 3285–3304. дои : 10.1093/нар/13.9.3285 . ISSN 0305-1048 . ПМК 341235 . ПМИД 2987881 .
- ^ Jump up to: а б с Ма, Вэньсю; Да, Ферхат; Ли, Чоли; Гулсой, Гунхан; Дэн, Синьсянь; Кук, Саванна; Хессон, Дженнифер; Кавано, Кристофер; Уэр, Кэрол Б.; Крумм, Антон; Шендюр, Джей; Блау, Карл Энтони; Дистече, Кристина М.; Ноубл, Уильям С.; Дуань, Чжицзюнь (январь 2015 г.). «Мелкомасштабные карты взаимодействия хроматина раскрывают цис-регуляторный ландшафт генов lincRNA человека» . Природные методы . 12 (1): 71–78. дои : 10.1038/nmeth.3205 . ISSN 1548-7105 . ПМЦ 4281301 . ПМИД 25437436 .
- ^ Jump up to: а б с Мифсуд, Борбала; Таварес-Кадете, Филипе; Янг, Элис Н.; Шугар, Роберт; Шенфельдер, Стефан; Феррейра, Лорен; Вингетт, Стивен В.; Эндрюс, Саймон; Грей, Уильям; Юэлс, Филип А.; Герман, Брэм; Хаппе, Скотт; Хиггс, Энди; ЛеПруст, Эмили ; Далее следует Джордж А.; Фрейзер, Питер; Ласкомб, Николас М.; Осборн, Кэмерон С. (июнь 2015 г.). «Картирование дальних контактов промотора в клетках человека с помощью захвата Hi-C с высоким разрешением» . Природная генетика . 47 (6): 598–606. дои : 10.1038/ng.3286 . ISSN 1546-1718 . ПМИД 25938943 . S2CID 6036717 .
- ^ Каллен, Кэтрин Э.; Кладде, Майкл П.; Сейфред, Марк А. (9 июля 1993 г.). «Взаимодействие между регионами регуляции транскрипции пролактина хроматина» . Наука . 261 (5118): 203–206. Бибкод : 1993Sci...261..203C . дои : 10.1126/science.8327891 . ПМИД 8327891 .
- ^ Чжоу, Юфань; Ченг, Сяолун; Ян, Ини; Ли, Тиан; Ли, Цзинвэй; Хуанг, Тим Х.-М.; Ван, Джунбай; Лин, Шили; Джин, Виктор X. (12 августа 2020 г.). «Моделирование и анализ данных Hi-C с помощью HiSIF выявляет характерные дистальные петли промотора» . Геномная медицина . 12 (1): 69. дои : 10.1186/s13073-020-00769-8 . ISSN 1756-994Х . ПМК 7425017 . ПМИД 32787954 .
- ^ Jump up to: а б с д и ж г час я Диас, Ноэлия; Крузе, Кай; Эрдманн, Табеа; Штайгер, Аннет М.; Отт, немецкий; Ленц, Георг; Вакерисас, Хуан М. (29 ноября 2018 г.). «Анализ конформации хроматина первичной ткани пациента с использованием метода Hi-C с низким входом» . Природные коммуникации . 9 (1): 4938. Бибкод : 2018NatCo...9.4938D . дои : 10.1038/s41467-018-06961-0 . ISSN 2041-1723 . ПМК 6265268 . ПМИД 30498195 .
- ^ Jump up to: а б с д де Соуза, Натали (сентябрь 2015 г.). «Микро-С-карты структуры генома» . Природные методы . 12 (9): 812. doi : 10.1038/nmeth.3575 . ISSN 1548-7105 . ПМИД 26554092 . S2CID 5765554 .
- ^ Jump up to: а б с д Берджесс, Даррен Дж. (июнь 2020 г.). «Хромосомная структура на микроуровне» . Обзоры природы Генетика . 21 (6): 337. doi : 10.1038/s41576-020-0243-y . ISSN 1471-0064 . ПМИД 32346116 . S2CID 216560645 .
- ^ Jump up to: а б Се, Цунг-Хан С.; Каттольо, Клаудия; Слободянюк Елена; Хансен, Андерс С.; Рандо, Оливер Дж.; Тиан, Роберт; Дарзак, Ксавье (7 мая 2020 г.). «Разрешение трехмерного ландшафта сворачивания связанного с транскрипцией хроматина млекопитающих» . Молекулярная клетка . 78 (3): 539–553.e8. doi : 10.1016/j.molcel.2020.03.002 . ISSN 1097-2765 . ПМЦ 7703524 . ПМИД 32213323 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо топор является тот нет бб до нашей эры др. быть парень Ладжуа, Брайан Р.; Деккер, Иов; Каплан, Ноам (январь 2015 г.). «Автостопом по анализу Hi-C: Практические рекомендации» . Методы . 72 : 65–75. дои : 10.1016/j.ymeth.2014.10.031 . ISSN 1046-2023 . ПМЦ 4347522 . ПМИД 25448293 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть Пал, Кустав; Форкато, Маттиа; Феррари, Франческо (20 декабря 2018 г.). «Hi-C анализ: от генерации данных к интеграции» . Биофизические обзоры . 11 (1): 67–78. дои : 10.1007/s12551-018-0489-1 . ISSN 1867-2450 . ПМК 6381366 . ПМИД 30570701 .
- ^ Jump up to: а б с Форкато, Маттиа; Биччато, Сильвио (2021). «Вычислительный анализ данных Hi-C». Захват конформации хромосомы . Методы молекулярной биологии. Том. 2157. Спрингер США. стр. 103–125. дои : 10.1007/978-1-0716-0664-3_7 . ISBN 978-1-0716-0663-6 . ПМИД 32820401 . S2CID 221219811 .
{{cite book}}:|journal=игнорируется ( помогите ) - ^ Jump up to: а б с д и ж г час я Гонг, Хайян; Ян, И; Чжан, Сичэн; Ли, Минхун; Чжан, Сяотун (1 января 2021 г.). «Применение Hi-C и другого анализа данных омики в исследованиях рака человека и дифференциации клеток» . Журнал вычислительной и структурной биотехнологии . 19 : 2070–2083. дои : 10.1016/j.csbj.2021.04.016 . ISSN 2001-0370 . ПМК 8086027 . ПМИД 33995903 .
- ^ Лэнгмид, Бен (декабрь 2010 г.). «Согласование считываний коротких последовательностей с помощью галстука-бабочки» . Современные протоколы в биоинформатике . 32 (1): Раздел 11.7. дои : 10.1002/0471250953.bi1107s32 . hdl : 2027.42/137758 . ISSN 1934-3396 . ПМК 3010897 . ПМИД 21154709 .
- ^ Ли, Хэн; Дурбин, Ричард (15 июля 2009 г.). «Быстрое и точное выравнивание короткого чтения с помощью преобразования Берроуза – Уиллера» . Биоинформатика . 25 (14): 1754–1760. doi : 10.1093/биоинформатика/btp324 . ПМК 2705234 . ПМИД 19451168 .
- ^ Ли, Хэн (2018). «Миникарта2: попарное выравнивание нуклеотидных последовательностей» . Биоинформатика . 34 (18): 3094–3100. doi : 10.1093/биоинформатика/bty191 . ПМК 6137996 . ПМИД 29750242 .
- ^ Jump up to: а б Имакаев Максим; Фуденберг, Джеффри; МакКорд, Рэйчел Паттон; Наумова, Наталья; Голобородько Антон; Ладжуа, Брайан Р.; Деккер, Иов; Мирный, Леонид А. (октябрь 2012 г.). «Итеративная коррекция данных Hi-C выявляет признаки организации хромосом» . Природные методы . 9 (10): 999–1003. дои : 10.1038/nmeth.2148 . ISSN 1548-7105 . ПМЦ 3816492 . ПМИД 22941365 .
- ^ Jump up to: а б Слуга Николас; Варокво, Нелль; Ладжуа, Брайан Р.; Виара, Эрик; Чен, Чун-Цзянь; Верт, Жан-Филипп; Слышал, Эдит; Деккер, Иов; Барийо, Эммануэль (1 декабря 2015 г.). «HiC-Pro: оптимизированный и гибкий конвейер для обработки данных Hi-C» . Геномная биология . 16 (1): 259. дои : 10.1186/s13059-015-0831-x . ISSN 1474-760X . ПМЦ 4665391 . ПМИД 26619908 .
- ^ Хван, Йи-Чии; Линь, Цзяо-Фэн; Валладарес, Отто; Маламон, Джон; Кукса, Павел П.; Чжэн, Ци; Грегори, Брайан Д.; Ван, Ли-Сан (15 апреля 2015 г.). «HIPPIE: высокопроизводительный конвейер идентификации взаимодействующих с промотором энхансерных элементов» . Биоинформатика . 31 (8): 1290–1292. doi : 10.1093/биоинформатика/btu801 . ISSN 1367-4803 . ПМЦ 4393516 . ПМИД 25480377 .
- ^ Вингетт, Стивен; Юэлс, Филип; Фурлан-Магарил, Майра; Нагано, Такаши; Шенфельдер, Стефан; Фрейзер, Питер; Эндрюс, Саймон (20 ноября 2015 г.). «HiCUP: конвейер для отображения и обработки данных Hi-C» . F1000Исследования . 4 : 1310. doi : 10.12688/f1000research.7334.1 . ISSN 2046-1402 . ПМК 4706059 . ПМИД 26835000 .
- ^ Серра, Франсуа; Бау, Давиде; Гудштадт, Майк; Кастильо, Дэвид; Филион, Гийом Ж.; Марти-Реном, Марк А. (19 июля 2017 г.). «Автоматический анализ и 3D-моделирование данных Hi-C с использованием TADbit выявляет структурные особенности цветов хроматина мух» . PLOS Вычислительная биология . 13 (7): e1005665. Бибкод : 2017PLSCB..13E5665S . дои : 10.1371/journal.pcbi.1005665 . ISSN 1553-7358 . ПМК 5540598 . ПМИД 28723903 .
- ^ Дюран, Нева С.; Шамим, Мухаммад С.; Махол, Идо; Рао, Сухас СП; Хантли, Мириам Х.; Ландер, Эрик С.; Эйден, Эрез Либерман (27 июля 2016 г.). «Соковыжималка предоставляет систему в один клик для анализа экспериментов Hi-C с петлевым разрешением» . Клеточные системы . 3 (1): 95–98. дои : 10.1016/j.cels.2016.07.002 . ISSN 2405-4712 . ПМЦ 5846465 . ПМИД 27467249 .
- ^ Рейфф, Сара Б.; Шредер, Эндрю Дж.; Кырли, Корай; Косоло, Андреа; Баккер, Клара; Ли, Сухён; Вейт, Александр Д.; Балашов Александр К.; Вицтум, Карл; Рончетти, Уильям; Питман, Кент М.; Джонсон, Джереми; Эмсен, Шеннон Р.; Керпеджиев, Петр; Абденнур, Незар; Имакаев Максим; Озтюрк, Серкан Утку; Чамоглу, Угур; Мирный, Леонид А.; Геленборг, Нильс; Алвер, Бурак Х.; Парк, Питер Дж. (2022). «Портал 4D-нуклеомных данных как ресурс для поиска и визуализации тщательно подобранных данных нуклеомы» . Природные коммуникации . 13 (1): 2365. Бибкод : 2022NatCo..13.2365R . дои : 10.1038/s41467-022-29697-4 . ПМК 9061818 . ПМИД 35501320 .
- ^ Ли, Хэн (2013). «Согласование считывания последовательностей, последовательностей клонирования и контигов сборки с помощью BWA-MEM». arXiv : 1303.3997 [ q-bio.GN ].
- ^ Чжан, Х.; Песня, Л.; Ван, X.; Ченг, Х.; Ван, К.; Мейер, Калифорния; Лю, Т.; Тан, М.; Алуру, С.; Юэ, Ф.; Лю, XS; Ли, Х. (2021). «Быстрое выравнивание и предварительная обработка профилей хроматина с помощью Chromap» . Природные коммуникации . 12 (1): 6566. Бибкод : 2021NatCo..12.6566Z . doi : 10.1038/s41467-021-26865-w . ПМЦ 8589834 . ПМИД 34772935 .
- ^ Форкато, Маттиа; Николетти, Кьяра; Пал, Кустав; Ливи, Кармен Мария; Феррари, Франческо; Биччато, Сильвио (июль 2017 г.). «Сравнение вычислительных методов анализа данных Hi-C» . Природные методы . 14 (7): 679–685. дои : 10.1038/nmeth.4325 . ISSN 1548-7105 . ПМЦ 5493985 . ПМИД 28604721 .
- ^ Нора, Эльфеж П.; Голобородько Антон; Валтон, Анн-Лора; Гибкус, Йохан Х.; Юберсон, Алек; Абденнур, Незар; Деккер, Иов; Мирный, Леонид А.; Брюно, Бенуа Г. (18 мая 2017 г.). «Направленная деградация CTCF отделяет локальную изоляцию хромосомных доменов от компартментализации генома» . Клетка . 169 (5): 930–944.e22. дои : 10.1016/j.cell.2017.05.004 . ISSN 0092-8674 . ПМК 5538188 . ПМИД 28525758 .
- ^ Ли, Ангшэн; Инь, Сяньчэнь; Сюй, Бинсян; Ван, Даньян; Хан, Чимин; Вэй, И; Дэн, Юн; Сюн, Инь; Чжан, Чжихуа (15 августа 2018 г.). «Декодирование топологически связанных доменов с данными Hi-C сверхнизкого разрешения с помощью структурной энтропии графа» . Природные коммуникации . 9 (1): 3265. Бибкод : 2018NatCo...9.3265L . дои : 10.1038/s41467-018-05691-7 . ISSN 2041-1723 . ПМК 6093941 . ПМИД 30111883 .
- ^ Саурия, Майкл Э.Г.; Тейлор, Джеймс (14 ноября 2017 г.). «QuASAR: Оценка качества воспроизводимости пространственного расположения в данных Hi-C» : 204438. doi : 10.1101/204438 . S2CID 90376810 .
{{cite journal}}: Для цитирования журнала требуется|journal=( помощь ) - ^ Рамирес, Фидель; Лингг, Томас; Тоскано, Сара; Лам, Кин Чунг; Георгиев, Пламен; Чунг, Хо-Рюн; Ладжуа, Брайан; де Вит, Эльцо; Чжан, Е; де Лаат, Воутер; Деккер, Иов; Манке, Томас; Ахтар, Асифа (1 октября 2015 г.). «Сайты с высоким сродством образуют сеть взаимодействия, способствующую распространению комплекса MSL по Х-хромосоме у дрозофилы» . Молекулярная клетка . 60 (1): 146–162. doi : 10.1016/j.molcel.2015.08.024 . ISSN 1097-2765 . ПМК 4806858 . ПМИД 26431028 .
- ^ Jump up to: а б Чжан, Ян; Ан, Лин; Сюй, Цзе; Чжан, Бо; Чжэн, В. Джим; Ху, Мин; Тан, Цзиджун; Юэ, Фэн (21 февраля 2018 г.). «Повышение разрешения данных Hi-C с помощью глубокой сверточной нейронной сети HiCPlus» . Природные коммуникации . 9 (1): 750. Бибкод : 2018NatCo...9..750Z . дои : 10.1038/s41467-018-03113-2 . ISSN 2041-1723 . ПМК 5821732 . ПМИД 29467363 .
- ^ Jump up to: а б с Кнопп, Пол; Синкхорн, Ричард (январь 1967 г.). «О неотрицательных матрицах и дважды стохастических матрицах» . Тихоокеанский математический журнал . 21 (2): 343–348. дои : 10.2140/pjm.1967.21.343 . ISSN 0030-8730 .
- ^ Курнак, Аксель; Мари-Нелли, Эрве; Марбути, Марсьяль; Кошул, Ромен; Моцциконаччи, Жюльен (30 августа 2012 г.). «Нормализация карты контактов хромосом» . БМК Геномика . 13 : 436. дои : 10.1186/1471-2164-13-436 . ISSN 1471-2164 . ПМЦ 3534615 . ПМИД 22935139 .
- ^ Найт, Филип; Руис, Даниэль (26 октября 2012 г.). «Быстрый алгоритм балансировки матрицы» . Журнал IMA численного анализа . 33 (3): 1029–1047. дои : 10.1093/imanum/drs019 .
- ^ Jump up to: а б с Раккоста, Самуэле; Либрицци, Фабио; Джаггер, Алистер М.; Ното, Розина; Марторана, Винченцо; Ломас, Дэвид А.; Ирвинг, Джеймс А.; Манно, Мауро (январь 2021 г.). «Концепции масштабирования в физике серпин-полимеров» . Материалы . 14 (10): 2577. Бибкод : 2021Mate...14.2577R . дои : 10.3390/ma14102577 . ISSN 1996-1944 гг . ПМЦ 8156723 . ПМИД 34063488 .
- ^ Jump up to: а б с Фуденберг, Джеффри; Мирный, Леонид А (1 апреля 2012 г.). «Структура хроматина высшего порядка: соединение физики и биологии» . Текущее мнение в области генетики и развития . 22 (2): 115–124. дои : 10.1016/j.где.2012.01.006 . hdl : 1721.1/103044 . ISSN 0959-437X . ПМЦ 3697851 . ПМИД 22360992 .
- ^ Хайнц, Свен; Беннер, Кристофер; Спанн, Натаниэль; Бертолино, Эрик; Линь, Инь С.; Ласло, Питер; Ченг, Джейсон X.; Мурре, Корнелис; Сингх, Хариндер; Гласс, Кристофер К. (28 мая 2010 г.). «Простые комбинации факторов транскрипции, определяющих происхождение, запускают цис-регуляторные элементы, необходимые для идентичности макрофагов и В-клеток» . Молекулярная клетка . 38 (4): 576–589. doi : 10.1016/j.molcel.2010.05.004 . ISSN 1097-2765 . ПМЦ 2898526 . ПМИД 20513432 .
- ^ Чжэн, Сяобин; Чжэн, Исянь (1 мая 2018 г.). «CscoreTool: быстрый анализ отсеков Hi-C с высоким разрешением» . Биоинформатика . 34 (9): 1568–1570. doi : 10.1093/биоинформатика/btx802 . ISSN 1367-4803 . ПМЦ 5925784 . ПМИД 29244056 .
- ^ де Вит, Эльцо (7 февраля 2020 г.). «TAD, как их называет вызывающий абонент» . Журнал молекулярной биологии . 432 (3): 638–642. дои : 10.1016/j.jmb.2019.09.026 . ISSN 0022-2836 . ПМИД 31654669 . S2CID 204918507 .
- ^ Наумова, Наталья; Имакаев Максим; Фуденберг, Джеффри; Чжан, Е; Ладжуа, Брайан Р.; Мирный, Леонид А.; Деккер, Иов (22 ноября 2013 г.). «Организация митотической хромосомы» . Наука . 342 (6161): 948–953. Бибкод : 2013Sci...342..948N . дои : 10.1126/science.1236083 . ISSN 0036-8075 . ПМК 4040465 . ПМИД 24200812 .
- ^ Jump up to: а б с Крейн, Эмили; Бянь, Цянь; МакКорд, Рэйчел Паттон; Ладжуа, Брайан Р.; Уиллер, Бэйли С.; Ралстон, Эдвард Дж.; Узава, Сатору; Деккер, Иов; Мейер, Барбара Дж. (июль 2015 г.). «Управляемое конденсином ремоделирование топологии Х-хромосомы во время дозовой компенсации» . Природа . 523 (7559): 240–244. Бибкод : 2015Natur.523..240C . дои : 10.1038/nature14450 . ISSN 1476-4687 . ПМЦ 4498965 . ПМИД 26030525 .
- ^ Jump up to: а б с Филиппова, Дарья; Патро, Роб; Дуггал, Гит; Кингсфорд, Карл (3 мая 2014 г.). «Идентификация альтернативных топологических доменов в хроматине» . Алгоритмы молекулярной биологии . 9 (1): 14. дои : 10.1186/1748-7188-9-14 . ISSN 1748-7188 . ПМК 4019371 . ПМИД 24868242 .
- ^ Шварцер, Вибке; Абденнур, Незар; Голобородько Антон; Пековская, Александра; Фуденберг, Джеффри; Ло-Ми, Янн; Фонсека, Нуно А; Хубер, Вольфганг; Херинг, Кристиан; Мирный, Леонид; Шпиц, Франсуа (2 ноября 2017 г.). «Два независимых способа организации хроматина, выявленные путем удаления когезина» . Природа . 551 (7678): 51–56. Бибкод : 2017Natur.551...51S . дои : 10.1038/nature24281 . ISSN 0028-0836 . ПМЦ 5687303 . ПМИД 29094699 .
- ^ Ян, Кун-Киу; Лу, Шаоке; Герштейн, Марк (24 июля 2017 г.). «MrTADFinder: подход, основанный на сетевой модульности, для идентификации топологически связанных доменов в нескольких разрешениях» . PLOS Вычислительная биология . 13 (7): e1005647. Бибкод : 2017PLSCB..13E5647Y . дои : 10.1371/journal.pcbi.1005647 . ISSN 1553-734X . ПМЦ 5546724 . ПМИД 28742097 .
- ^ Нортон, Хайди К.; Эмерсон, Дэниел Дж.; Хуанг, Харви; Ким, Джези; Титус, Кейтлин Р.; Гу, Ши; Бассетт, Даниэль С.; Филлипс-Креминс, Дженнифер Э. (февраль 2018 г.). «Обнаружение иерархического сворачивания генома с помощью сетевой модульности» . Природные методы . 15 (2): 119–122. дои : 10.1038/nmeth.4560 . ISSN 1548-7105 . ПМК 6029251 . ПМИД 29334377 .
- ^ Малик, Лараиб; Патро, Роб (сентябрь 2019 г.). «Прогнозирование богатой структуры хроматина на основе данных Hi-C» . Транзакции IEEE/ACM по вычислительной биологии и биоинформатике . 16 (5): 1448–1458. дои : 10.1109/TCBB.2018.2851200 . ISSN 1557-9964 . ПМИД 29994683 . S2CID 54563346 .
- ^ Jump up to: а б Оу, Хорнг Д.; Фан, Себастьен; Диринк, Томас Дж.; Тор, Андреа; Эллисман, Марк Х.; О'Ши, Клода К. (28 июля 2017 г.). «ChromEMT: визуализация трехмерной структуры хроматина и уплотнения интерфазных и митотических клеток» . Наука . 357 (6349): eaag0025. дои : 10.1126/science.aag0025 . ISSN 0036-8075 . ПМЦ 5646685 . ПМИД 28751582 .
- ^ Jump up to: а б Нагано, Такаши; Люблинг, Янив; Варнаи, Чилла; Дадли, Кармель; Люнг, Винг; Баран, Яэль; Мендельсон Коэн, Нетта; Вингетт, Стивен; Фрейзер, Питер; Танай, Амос (июль 2017 г.). «Динамика клеточного цикла хромосомной организации при разрешении одной клетки» . Природа . 547 (7661): 61–67. Бибкод : 2017Natur.547...61N . дои : 10.1038/nature23001 . ISSN 1476-4687 . ПМЦ 5567812 . ПМИД 28682332 .
- ^ Бинту, Богдан; Матео, Лесли Дж.; Су, Джун-Хан; Синнотт-Армстронг, Николас А.; Паркер, Мирэ; Кинрот, Сон; Ямая, Кей; Беттигер, Алистер Н.; Чжуан, Сяовэй (26 октября 2018 г.). «Отслеживание хроматина со сверхвысоким разрешением выявляет домены и кооперативные взаимодействия в отдельных клетках» . Наука . 362 (6413): eaau1783. Бибкод : 2018Sci...362.1783B . дои : 10.1126/science.aau1783 . ISSN 0036-8075 . ПМК 6535145 . ПМИД 30361340 .
- ^ Jump up to: а б Ю, Мяо; Рен, Бинг (6 октября 2017 г.). «Трехмерная организация геномов млекопитающих» . Ежегодный обзор клеточной биологии и биологии развития . 33 (1): 265–289. doi : 10.1146/annurev-cellbio-100616-060531 . ISSN 1081-0706 . ПМЦ 5837811 . ПМИД 28783961 .
- ^ Ниакан, Кэти К.; Хан, Джиннуо; Педерсен, Роджер А.; Саймон, Карлос; Пера, Рене А. Рейхо (1 марта 2012 г.). «Преимплантационное развитие эмбриона человека» . Разработка . 139 (5): 829–841. дои : 10.1242/dev.060426 . ISSN 0950-1991 . ПМК 3274351 . ПМИД 22318624 .
- ^ Jump up to: а б с Ду, Чжэн, Хуэй, Ма, Жуй, Чжан, Сянлинь; Ван, Цюцзюнь, Ма, Цзин, Сюй; Кэ; Ван, Майкл К.; Гао, Диксон, Джесси Р.; Ван, Цзяньян; Се, Вэй (июль 2017 г.) . . Природа . 547 ): Бибкод : 2017Natur.547..232D . doi : / . ISSN 1476-4687 . nature23263 7662 . ( 10.1038 232–235
- ^ Jump up to: а б с Кэ, Ювэнь, Сюэпэн; Фэн, Лю, Чжэньбо; Ли, Фанчжэнь; Чэнь, Хаоцзе; Хуан, Лю, Цзян (13 июля 2017 г.). перепрограммирование во время эмбриогенеза млекопитающих» . гамет структуры хроматина зрелых 3D Сюй, Сяоцуй ; и структурное « .cell.2017.06.029 . ISSN 0092-8674 . PMID 28709003. S2CID 23974814 .
- ^ Диамент, Алон; Туллер, Тамир (1 июня 2019 г.). «Моделирование трехмерной геномной организации в эволюции и патогенезе» . Семинары по клеточной биологии и биологии развития . 90 : 78–93. дои : 10.1016/j.semcdb.2018.07.008 . ISSN 1084-9521 . ПМИД 30030143 . S2CID 51704135 .
- ^ Jump up to: а б Виетри, Маттео; Баррингтон, Кристофер; Хендерсон, Стивен; Эрнст, Кристина; Одом, Дункан; Танай, Амос; Хаджур, Сузана (26 февраля 2015 г.). «Сравнительный Hi-C показывает, что CTCF лежит в основе эволюции архитектуры хромосомного домена» . Отчеты по ячейкам . 10 (8): 1297–1309. дои : 10.1016/j.celrep.2015.02.004 . ISSN 2211-1247 . ПМЦ 4542312 . ПМИД 25732821 .
- ^ Диамент, Алон; Пинтер, Рон Ю.; Туллер, Тамир (16 декабря 2014 г.). «Трёхмерная геномная организация эукариот сильно коррелирует с экспрессией и функцией использования кодонов» . Природные коммуникации . 5 (1): 5876. Бибкод : 2014NatCo...5.5876D . дои : 10.1038/ncomms6876 . ISSN 2041-1723 . ПМИД 25510862 .
- ^ Ибн-Салем, Йонас; Муро, Энрике М.; Андраде-Наварро, Мигель А. (9 января 2017 г.). «Совместная регуляция генов паралогов в трехмерной архитектуре хроматина» . Исследования нуклеиновых кислот . 45 (1): 81–91. дои : 10.1093/нар/gkw813 . ISSN 0305-1048 . ПМК 5224500 . ПМИД 27634932 .
- ^ Диамент, Алон; Туллер, Тамир (5 мая 2017 г.). «Отслеживание эволюции трехмерной организации генов демонстрирует ее связь с фенотипической дивергенцией» . Исследования нуклеиновых кислот . 45 (8): 4330–4343. дои : 10.1093/нар/gkx205 . ISSN 0305-1048 . ПМЦ 5416853 . ПМИД 28369658 .
- ^ Бонев, Боян; Кавалли, Джакомо (ноябрь 2016 г.). «Организация и функции 3D-генома» . Обзоры природы Генетика . 17 (11): 661–678. дои : 10.1038/nrg.2016.112 . ISSN 1471-0064 . ПМИД 27739532 . S2CID 31259189 .
- ^ Чемберс, Эмили В.; Бикмор, Венди А.; Семпл, Колин А. (4 апреля 2013 г.). «Дивергенция структуры хроматина высшего порядка млекопитающих связана с локусами развития» . PLOS Вычислительная биология . 9 (4): e1003017. Бибкод : 2013PLSCB...9E3017C . дои : 10.1371/journal.pcbi.1003017 . ISSN 1553-7358 . ПМК 3617018 . ПМИД 23592965 .
- ^ Jump up to: а б с Клотген, Андреас; Тандапани, Паланираджа; Нциахристос, Панайотис; Гебрехристос, Йохана; Номику, София; Лазарис, Харалампос; Чен, Сюйфэн; Ху, Хай; Бакоянни, София; Ван, Цзинцзин; Фу, Йи; Боккалатте, Франческо; Чжун, Хуа; Пайетта, Элизабет; Тримарчи, Томас; Чжу, Исин; Ван Влиерберге, Питер; Ингирами, Джорджио Г.; Лайоннет, Тимоти; Айфантис, Яннис; Циригос, Аристотелис (апрель 2020 г.). «Трехмерные ландшафты хроматина при остром Т-клеточном лимфобластном лейкозе» . Природная генетика . 52 (4): 388–400. дои : 10.1038/s41588-020-0602-9 . ISSN 1546-1718 . ПМЦ 7138649 . ПМИД 32203470 .
- ^ Jump up to: а б с Фанг, Селестия; Ван, Чжэньцзя; Хан, Цуйцзюань; Сафгрен, Стефани Л.; Хелмин, Кэтрин А.; Адельман, Эммали Р.; Серафин, Валентина; Бассо, Джузеппе; Иген, Кайл П.; Гаспар-Майя, Александр; Фигероа, Мария Э.; Певец Бенджамин Д.; Ратан, Аакрош; Нциахристос, Панайотис; Занг, Чунчжи (15 сентября 2020 г.). «Специфическое для рака связывание CTCF способствует онкогенной дисрегуляции транскрипции» . Геномная биология . 21 (1): 247. дои : 10.1186/s13059-020-02152-7 . ISSN 1474-760X . ПМЦ 7493976 . ПМИД 32933554 .