Окисление, катализируемое оксоаммонием
Реакции окисления, катализируемые оксоаммонием, включают преобразование органических субстратов в более сильно окисленные материалы под действием разновидностей N-оксоаммония . Нитроксиды также можно использовать в каталитических количествах в присутствии стехиометрического количества терминального окислителя. [ 1 ] Используемые нитроксидные радикалы представляют собой либо 2,2,6,6-тетраметилпиперидин-1-оксил (ТЕМПО), либо его производные.
(1)

Механизм и стереохимия
[ редактировать ]Одноэлектронное окисление нитроксида приводит к образованию высокоэлектрофильной разновидности оксоаммония, которая служит активным окислителем. [ 2 ] Нитроксид можно использовать в качестве катализатора в сочетании с более дешевыми стехиометрическими окислителями, такими как гипохлорит натрия. [ 3 ] или бис(ацетокси)иодбензол (BAIB). [ 4 ]
В нейтральных или слабокислых условиях (например, в присутствии силикагеля) окисление происходит за счет первоначальной водородной связи между гидроксильной группой и азотом оксоаммония с последующим согласованным переносом протона и отрывом гидрида. [ 5 ] Необходимость образования водородных связей подтверждается низкой реакционной способностью β-алкокси- и β-аминоспиртов, которые обладают конкурентной внутримолекулярной водородной связью. Механизм окисления в слабоосновных (пиридиновых) условиях аналогичен, за исключением того, что пиридин нейтрализует гидроксиаммониевые соединения, и этот промежуточный продукт «соразмеряется» с оксоаммониевой солью, образуя нитроксидные радикалы и соли пиридиния (см. уравнение (3) ниже). Поскольку в этой реакции расходуются основание и активный окислитель, в слабоосновных условиях необходимы два эквивалента основания и окислителя. Единый механизм при нейтральных и базовых условиях представлен в недавней статье. [ 6 ] Авторы представляют комплексный анализ ряда процессов окисления, опосредованных оксоаммониевыми солями.
(2)

В сильноосновных условиях депротонированный субстрат реагирует с частицами N-оксиаммония. Может произойти атака алкоксида субстрата как на азот, так и на кислород, хотя считается, что первый действует на основании наблюдений за окислением N-алкоксиаминов (которые, предположительно, протекают через промежуточное соединение 1 ). [ 7 ] Сопропорционирование восстановленного продукта (гидроксиламина) с ионом оксоаммония конкурирует с окислением; таким образом, часто требуется избыток окислителя.
(3)

В окислении, катализируемом нитроксидом, в качестве активного окислителя используются промежуточные соединения N-оксоаммония. Механизм окисления нитроксидного радикала зависит от используемого терминального окислителя. Двухэлектронные окислители, такие как NaOCl, способны напрямую превращать нитроксиды в оксоаммоний.
(4)

Одноэлектронные окислители, такие как медь (II), действуют по более сложному механизму, в котором в качестве терминального окислителя используется дикислород. [ 8 ] Медь(II) окисляет четыре эквивалента нитроксида до оксоаммония, два эквивалента которого (синий) реагируют со спиртами с образованием карбонильных соединений. Два других эквивалента оксоаммония (красный) подвергаются сопропорционированию с образованием нитрокси-радикалов (розовый). Наконец, дикислород повторно окисляет четыре эквивалента меди(I) обратно в медь(II). В целом, одна молекула дикислорода опосредует окисление двух эквивалентов спирта с образованием двух эквивалентов воды.
(5)

Стереоселективные варианты
[ редактировать ]Энантиоселективное окисление обычно представляет собой либо кинетическое разделение хиральных спиртов, либо реакции десимметризации. Этим окислениям можно способствовать за счет использования хиральных нитроксидных радикалов в каталитическом режиме. Хорошим примером является кинетическое разрешение рацемического 1-фенилэтанола. [ 9 ] С другой стороны, процессы окислительной десимметризации с использованием оксоаммониевых окислителей относительно редки. [ 10 ]
(6)

Объем
[ редактировать ]Окисление с использованием оксоаммониевых солей может проводиться как в стехиометрическом, так и в каталитическом режиме в кислых или основных условиях. В этом разделе описаны наиболее часто используемые условия стехиометрического и каталитического окисления спиртов до карбонильных соединений оксоаммониевыми солями. Хотя с помощью TEMPO можно окислить самые разные спирты, иногда происходит конкурентное окисление более богатых электронами функциональных групп. Кроме того, селективность сайта окисления полиолов может различаться в зависимости от используемых условий.
Стехиометрические окисления
[ редактировать ]В слабокислых или нейтральных условиях соли оксоаммония, такие как соль Боббита, окисляют аллильные, бензильные, [ 11 ] пропаргиловый, [ 12 ] или алифатические спирты к соответствующим альдегидам или кетонам. Вторичные спирты реагируют быстрее, чем первичные, хотя селективность низкая. Удобный протокол эксперимента позволяет повторно использовать оксоаммониевую соль. [ 12 ]
(7)

Амины, бензиловые эфиры и алкены окисляются быстрее, чем неактивированные спирты; таким образом, селективное стехиометрическое окисление неактивированных спиртов в присутствии этих функциональных групп невозможно. [ 13 ] Спирты с β-азотными или β-кислородными заместителями медленно реагируют в кислой среде. [ 12 ] В этих условиях аллиловые и бензиловые спирты могут избирательно окисляться. [ 13 ]
(8)

В основных условиях необходимы два эквивалента окислителя из-за конкурентного пропорционального соотношения между восстановленным нитроксидом и непрореагировавшим оксоаммонием (см. уравнение (3) выше). В качестве основания обычно используют пиридин. Это наиболее распространенные условия окисления нитроксидов в стехиометрическом режиме.
(9)

Третичные аллильные спирты также могут быть стехиометрически окислены оксоаммониевыми солями до енонов в варианте реакции Баблера-Даубена . [ 14 ]
Каталитическое окисление
[ редактировать ]Каталитическое окисление оксоаммония можно облегчить, используя гипохлорит натрия в качестве терминального окислителя. Для продолжения реакции pH необходимо поддерживать ниже 10, используя буфер. Активным окислителем нитроксида является гипобромит-анион; следовательно, бромид калия используется в качестве добавки. [ 3 ] Эпимеризация α-стереогенных центров в карбонилсодержащих продуктах не происходит.
(10)

Использование хлоритов в качестве терминальных окислителей в сочетании как с гипохлоритами, так и с ТЕМПО дает карбоновые кислоты без побочных продуктов хлорирования. [ 15 ] Реакцию обычно проводят в две стадии в одном и том же котле: осуществляют частичное окисление ТЕМПО и гипохлоритом, затем для завершения окисления добавляют хлорит. Наблюдается только первичное окисление спирта. В сочетании с дигидроксилированием Шарплесса этот метод можно использовать для получения энантиочистых α-гидроксикислот. [ 16 ]
(11)

Существенным ограничением обоих вышеупомянутых методов является несовместимость со свободными амино- или алкеновыми группами, которые подвергаются конкурентному окислению. Использование бис(ацетокси)иодбензола (БАИБ) в качестве терминального окислителя позволяет избежать этой проблемы. BAIB не способен напрямую окислять нитроксидный радикал, и считается, что первоначальное образование оксоаммония происходит из-за диспропорционирования, катализируемого кислотой. Затем BAIB может окислить полученный гидроксиламин до оксоаммониевой соли. Хотя реакция проводится в кислых условиях (уксусная кислота является побочным продуктом и ее часто добавляют для облегчения диспропорционирования), селективность первичного окисления спирта является значительной. [ 4 ] В этих условиях переносятся чувствительные к основаниям функциональные группы, такие как эпоксиды. [ 17 ]
(12)
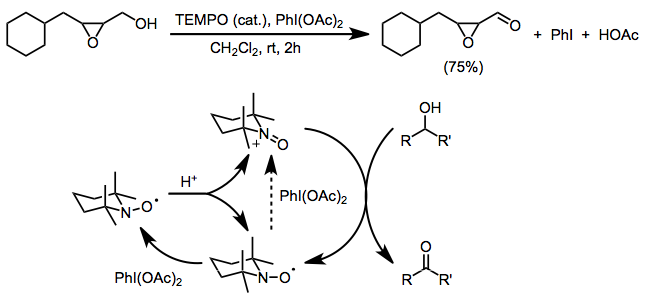
Другие двухэлектронные концевые окислители, используемые с TEMPO, включают mCPBA (предпочтительно вторичное окисление, хотя могут возникать побочные реакции), [ 18 ] N-хлорсукцинимид, [ 19 ] и Оксон. [ 20 ]
Медь(II) как в виде свободной хлоридной соли, так и в виде комплекса с бидентатными лигандами окисляет ТЕМПО до его оксоаммониевой соли. Конечным окислителем в этих реакциях является воздух. [ 21 ] Неясно, окисляет ли воздух медь(I) до меди(II) или окисление спирта частично опосредовано медью, а воздух окисляет образующийся гидроксиламин обратно в оксоаммониевую соль. Первое происходит во время процесса Вакера , но второе объясняет, почему комплексы меди и некоторых других металлов способны окислять спирты в сочетании с ТЕМПО.
(13)

Сравнение с родственными методами
[ редактировать ]Активированный диоксид марганца , окисляющий аллиловые и бензиловые спирты, дешевле ТЕМПО и прост в эксплуатации. [ 22 ] Реагенты на основе хрома, такие как хлорхромат пиридиния, также могут использоваться для преобразования спиртов в карбонильные соединения; хотя стехиометрическое образование отходов хрома является недостатком. [ 23 ] Окисления с использованием диметилсульфоксида , такие как реакции Сверна и Моффата , не включают тяжелые металлы и окисляют широкий спектр субстратов. [ 24 ] Окисление оксоаммония предпочтительнее методов ДМСО для реакций диолов и ацетиленовых спиртов. Периодинан Десс-Мартена — высокоселективный, мягкий окислитель спиртов, основными недостатками которого являются трудности получения и безопасность. [ 25 ]
Ссылки
[ редактировать ]- ^ Боббитт, Дж. М .; Брукнер, К.; Мербух, Н. Орг. Реагировать. 2009 , 74 , 103. два : 10.1002/0471264180.or074.02
- ^ Merbouh, N.; Bobbitt, J. M.; Brückner, C. J. Org. Chem. 2004 , 69 , 5116.
- ^ Jump up to: а б Шелдон, РА ; Арендс, IWCE; тен Бринк, Дж.Дж.; Дейксман, А. Акк. хим. Рез. 2002 , 35 , 774. два : 10.1021/ar010075n
- ^ Jump up to: а б Де Мико, А.; Маргарита, Р.; Спикеры, Л.; Епископы, А.; Пианкателли, GJ Org. хим. 1997 , 62 , 6974.
- ^ Бэйли, WF; Боббитт, Дж. М.; Виберг, KB J. Org. хим. 2007 , 72 , 4504.
- ^ Хэмлин, штат Калифорния; Келли, CB; Овиан, Дж. М.; Уайлс, Р.Дж.; Тилли, Эл Джей; Ледбитер, штат Нью-Йорк, J. Org. хим. 2015 , 80 , 8150.
- ^ Семмельхак, МФ; Шмид, ЧР; Кортес, Д.А. Тетраэдр Летт. 1986 , 27 , 1119.
- ^ Семмельхак, МФ; Шмид, ЧР; Кортес, Д.А.; Чоу, C.S.J. Am. хим. Соц. 1984 , 106 , 3374.
- ^ Рыхновский, С.Д.; Маклернон, ТЛ; Раджапаксе, HJ Org. хим. 1996 , 61 , 1194.
- ^ , Ю.; Куробоши, Lett Tetrahedron М. Танака, Х.; Каваками .
- ^ .; Шиихаши, С . .; MJ Org Миядзава, Т
- ^ Jump up to: а б с Боббитт, JMJ Org. хим. 1998 , 63 , 9367.
- ^ Jump up to: а б Боббитт, Дж. М.; Мербух, Н. Орг. Синтез. 2005 , 82 , 80.>
- ^ Сибуя, Масатоши; Томизава, Масаки; Ивабути, Ёсихару (2008). «Окислительная перегруппировка третичных аллиловых спиртов с использованием оксоаммониевых солей» . Журнал органической химии . 73 (12): 4750–4752. дои : 10.1021/jo800634r . ISSN 0022-3263 . ПМИД 18500838 .
- ^ Песня, ZJ; Чжао, М.; Десмонд, Р.; Девайн, П.; Шэн, DM; Тиллер, Р.; Фрей, Л.; Хейд, Р.; Сюй, Ф.; Фостер, Б.; Ли, Дж.; Ример, Р.; Воланте, Р.; Грабовский, EJJ; Доллинг, США; Рейдер, П.Дж.; Окада, С.; Като, Ю.; Мано, EJ Org. хим. 1999 , 64 , 9658.
- ^ Шарплесс, КБ; Амберг, В.; Беннани, ЮЛ; Криспино, Джорджия; Хартунг, Дж.; Чон, Канзас; Квонг, HL; Морикава, К.; Ван, З.М.; Сюй, Д.; Чжан, XL J. Org. хим. 1992 , 57 , 2768.
- ^ Мичо, А.; Маргарита, Р.; Спикеры, Л.; Епископы, А.; Пианкателли, GJ Org. хим. 1997 , 62 , 6974.
- ^ Ганем, BJ Org. хим. 1975 , 40 , 1998.
- ^ Эйнхорн, Дж.; Эйнхорн, К.; Ратайчак, Ф.; Пьер, Ж.-Л. Ж. Орг. хим. 1996 , 61 , 7452.
- ^ Болм, К.; Магнус, А.С.; Хильдебранд, JP Org. Летт. 2000 , 2 , 1173.
- ^ Шелдон, РА ; Арендс, консультант IWCE. Синтез. Катал. 2004 , 346 , 1051.
- ^ Тейлор, RJK; Рид, М.; Фут, Дж.; Raw, SA Акк. хим. Рез. 2005 , 38 , 851.
- ^ Луццио, FA Org. Реагировать. 1998 , 53 , 1.
- ^ Тидвелл, TT Org. Реагировать. 1990 , 39 , 297.
- ^ Десс, Д.Б.; Мартин, JCJ Am. хим. Соц. 1991 , 113 , 7277.