БЛАТ (биоинформатика)
| Разработчик(и) | Джим Кент , UCSC |
|---|---|
| Репозиторий | |
| Тип | Инструмент биоинформатики |
| Лицензия | бесплатно для некоммерческого использования , коммерческое использование , исходный код доступен |
| Веб-сайт | геном |
BLAT ( BLAST -подобный инструмент выравнивания) — это парного выравнивания последовательностей алгоритм , который был разработан Джимом Кентом ( из Калифорнийского университета в Санта-Крус UCSC) в начале 2000-х годов для помощи в сборке и аннотировании генома человека . [ 1 ] Он был разработан в первую очередь для сокращения времени, необходимого для сопоставления миллионов считываний генома мыши и меток экспрессированных последовательностей с последовательностью генома человека. Инструменты выравнивания того времени не были способны выполнять эти операции таким образом, чтобы обеспечить регулярное обновление сборки генома человека. По сравнению с ранее существовавшими инструментами, BLAT работал примерно в 500 раз быстрее при выполнении выравнивания мРНК / ДНК и примерно в 50 раз быстрее при выравнивании белков /белков. [ 1 ]
Обзор
[ редактировать ]BLAT — это один из множества алгоритмов, разработанных для анализа и сравнения биологических последовательностей, таких как ДНК, РНК и белки, с основной целью определения гомологии для выявления биологической функции геномных последовательностей. [ 2 ] Не гарантируется, что будет найдено математически оптимальное выравнивание между двумя последовательностями, как в классическом случае Нидлмана-Вунша. [ 3 ] и Смит-Уотерман [ 4 ] динамического программирования алгоритмы делают это; скорее, он сначала пытается быстро обнаружить короткие последовательности, которые с большей вероятностью будут гомологичными, а затем выравнивает и дополнительно расширяет гомологичные области. Это похоже на эвристику BLAST. [ 5 ] [ 6 ] семейство алгоритмов, но каждый инструмент пытался решить проблему своевременного и эффективного выравнивания биологических последовательностей, пробуя различные алгоритмические методы. [ 2 ] [ 7 ]
Использование БЛАТ
[ редактировать ]BLAT можно использовать для выравнивания последовательностей ДНК, а также последовательностей белков и транслируемых нуклеотидов (мРНК или ДНК). Он предназначен для наилучшей работы с последовательностями с большим сходством. Поиск ДНК наиболее эффективен для приматов, а поиск белков — для наземных позвоночных. [ 1 ] [ 8 ] Кроме того, запросы белков или транслированных последовательностей более эффективны для выявления отдаленных совпадений и межвидового анализа, чем запросы последовательностей ДНК. [ 9 ] Типичные варианты использования BLAT включают следующее:
- Выравнивание нескольких последовательностей мРНК на сборке генома для определения их геномных координат; [ 10 ]
- Выравнивание последовательности белка или мРНК одного вида с базой данных последовательностей другого вида для определения гомологии. При условии, что эти два вида не слишком расходятся, межвидовое выравнивание обычно эффективно с помощью BLAT. Это возможно, поскольку BLAT не требует идеальных совпадений, а скорее допускает несоответствия в выравниваниях; [ 11 ]
- BLAT можно использовать для выравнивания двух белковых последовательностей. Однако это не лучший инструмент для такого типа выравнивания. BLASTP, инструмент Standard Protein BLAST , более эффективен при выравнивании белков; [ 1 ]
- Определение распределения экзонных и интронных областей гена; [ 9 ] [ 10 ]
- Обнаружение членов семейства генов по конкретному генному запросу; [ 9 ] [ 10 ]
- Отображение белково-кодирующей последовательности конкретного гена. [ 9 ] [ 10 ]
BLAT предназначен для поиска совпадений между последовательностями длиной не менее 40 оснований, которые имеют идентичность нуклеотидов ≥95% или идентичность транслируемых белков ≥80%. [ 9 ] [ 10 ]
Процесс
[ редактировать ]BLAT используется для поиска областей в целевой геномной базе данных, которые похожи на исследуемую последовательность запроса. Общий алгоритмический процесс, которому следует BLAT, аналогичен BLAST в том, что он сначала ищет короткие сегменты в базе данных и запрашивает последовательности, которые имеют определенное количество совпадающих элементов. Эти начальные числа выравнивания затем расширяются в обоих направлениях последовательностей, чтобы сформировать пары с высокой оценкой. [ 12 ] Однако BLAT использует другой подход к индексированию, чем BLAST, который позволяет ему быстро сканировать очень большие базы данных генома и белков на предмет сходства с последовательностью запроса. Это достигается за счет хранения в памяти индексированного списка ( хеш-таблицы ) целевой базы данных, что значительно сокращает время, необходимое для сравнения последовательностей запросов с целевой базой данных. Этот индекс строится путем взятия координат всех непересекающихся k-меров (слов с k буквами) в целевой базе данных, за исключением часто повторяющихся k-меров. Затем BLAT создает список всех перекрывающихся k-меров из последовательности запроса и ищет их в целевой базе данных, создавая список совпадений, в которых между последовательностями есть совпадения. [ 1 ] (Рисунок 1 иллюстрирует этот процесс).
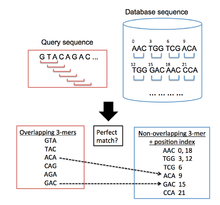
Этап поиска
[ редактировать ]Для поиска гомологичных регионов-кандидатов используются три различные стратегии:
- Первый метод требует одного идеального совпадения между последовательностями запроса и базы данных, т.е. два k-мерных слова абсолютно одинаковы. Этот подход не считается самым практичным. Это связано с тем, что для достижения высокого уровня чувствительности необходим небольшой размер k-мера, но это увеличивает количество ложноположительных совпадений, тем самым увеличивая количество времени, затрачиваемое на этап выравнивания алгоритма. [ 1 ]
- Второй метод допускает хотя бы одно несовпадение между двумя k-мерными словами. Это уменьшает количество ложных срабатываний, обеспечивая большие размеры k-меров, обработка которых требует меньше вычислительных затрат, чем те, которые получены с помощью предыдущего метода. Этот метод очень эффективен при идентификации небольших гомологичных областей. [ 1 ]
- Третий метод требует наличия нескольких идеальных совпадений, находящихся в непосредственной близости друг от друга. Как показывает Кент, [ 1 ] это очень эффективный метод, позволяющий учитывать небольшие вставки и делеции в гомологичных регионах.
При выравнивании нуклеотидов BLAT использует третий метод, требующий двух идеальных совпадений слов размером 11 (11-меров). При выравнивании белков версия BLAT определяет используемую методологию поиска: когда используется версия клиент/сервер, BLAT ищет три идеальных совпадения 4-меров; когда используется автономная версия, BLAT ищет один идеальный 5-мер между последовательностями запроса и базы данных. [ 1 ]
БЛАТ против BLAST
[ редактировать ]Некоторые различия между BLAT и BLAST описаны ниже:
- BLAT индексирует базу данных генома/белка, сохраняет индекс в памяти, а затем сканирует последовательность запросов на наличие совпадений. BLAST, с другой стороны, создает индекс последовательностей запросов и ищет совпадения в базе данных. [ 1 ] Вариант BLAST под названием MegaBLAST индексирует 4 базы данных для ускорения выравнивания. [ 9 ]
- BLAT может расширяться на несколько идеальных и почти идеальных совпадений (по умолчанию — 2 идеальных совпадения длиной 11 для поиска нуклеотидов и 3 идеальных совпадения длиной 4 для поиска белков), тогда как BLAST расширяется только тогда, когда одно или два совпадения встречаются близко друг к другу. [ 1 ] [ 9 ]
- BLAT соединяет каждую гомологичную область между двумя последовательностями в одно более крупное выравнивание, в отличие от BLAST, который возвращает каждую гомологичную область как отдельное локальное выравнивание. Результатом BLAST является список экзонов , каждое из которых простирается сразу за конец экзона. Однако BLAT правильно размещает каждое основание мРНК в геноме, используя каждое основание только один раз, и может использоваться для идентификации границ интрон -экзон (т.е. сайтов сплайсинга ). [ 1 ] [ 13 ]
- BLAT менее чувствителен, чем BLAST. [ 2 ]
Использование программы
[ редактировать ]BLAT можно использовать как веб-серверно-клиентскую программу, так и как отдельную программу. [ 9 ]
Сервер-клиент
[ редактировать ]Доступ к веб-приложению BLAT можно получить на сайте биоинформатики генома UCSC. [ 8 ] Построение индекса — относительно медленная процедура. Таким образом, каждая сборка генома, используемая веб-BLAT, связана с сервером BLAT, чтобы иметь предварительно вычисленный индекс, доступный для выравнивания. Эти веб-серверы BLAT хранят индекс в памяти, чтобы пользователи могли вводить последовательности запросов. [ 11 ]
После загрузки/вставки последовательности запроса в поле поиска пользователь может выбрать различные параметры, например, геном какого вида выбрать (в настоящее время доступно более 50 видов) и версию сборки этого генома (например, геном человека). имеет четыре сборки на выбор), тип запроса (т. е. относится ли последовательность к ДНК, белку и т. д.) и настройки вывода (т. е. как сортировать и визуализировать выходные данные). Затем пользователь может запустить поиск, отправив запрос или используя поиск BLAT «Мне повезет». [ 8 ]
Бхагват и др. [ 9 ] предоставить пошаговые протоколы использования BLAT для:
- Сопоставить последовательность мРНК/кДНК с геномной последовательностью;
- Сопоставьте последовательность белка с геномом;
- Выполните поиск гомологии.
Вход
[ редактировать ]BLAT может обрабатывать длинные последовательности баз данных, однако он более эффективен с короткими последовательностями запросов, чем с длинными последовательностями запросов. Кент [ 1 ] рекомендует максимальную длину запроса 200 000 баз. Браузер UCSC ограничивает последовательность запросов менее чем 25 000 букв (т. е. нуклеотидов ) для поиска ДНК и менее 10 000 букв (т. е. аминокислот ) для поиска белков и транслируемых последовательностей. [ 8 ]

Геном поиска BLAT, доступный на веб-сайте UCSC, принимает последовательности запросов в виде текста (вырезанные и вставленные в поле запроса) или загруженные в виде текстовых файлов. Поисковый геном BLAT может одновременно принимать несколько последовательностей одного типа, максимум до 25. Для нескольких последовательностей общее количество нуклеотидов не должно превышать 50 000 для поиска ДНК или 25 000 букв для поиска белков или транслируемых последовательностей. Пример поиска в целевой базе данных с помощью последовательности запроса ДНК показан на рисунке 2.
Выход
[ редактировать ]Поиск BLAT возвращает список результатов, упорядоченных в порядке убывания оценки. Возвращается следующая информация: оценка выравнивания, область последовательности запроса, которая соответствует последовательности базы данных, размер последовательности запроса, уровень идентичности в процентах от выравнивания, а также хромосома и положение, в котором находится последовательность запроса. карты в. [ 9 ] Бхагват и др. [ 9 ] опишите, как рассчитываются показатели BLAT «Оценка» и «Идентичность».
Для каждого результата поиска пользователю предоставляется ссылка на браузер генома UCSC, чтобы он мог визуализировать выравнивание хромосом. Это главное преимущество веб-BLAT по сравнению с автономным BLAT. Пользователь может получить биологическую информацию, связанную с выравниванием, например информацию о гене, которому может соответствовать запрос. [ 9 ] Пользователю также предоставляется ссылка для просмотра соответствия последовательности запроса сборке генома. Совпадения между запросом и сборкой генома выделены синим цветом, а границы выравниваний светлее. Эти границы экзонов указывают сайты сплайсинга. [ 8 ] [ 9 ] Результат поиска «Мне повезет» возвращает выравнивание с наивысшим баллом для первой последовательности запросов на основе параметра сортировки вывода, выбранного пользователем. [ 8 ]
Автономный
[ редактировать ]Автономный BLAT больше подходит для пакетного запуска и более эффективен, чем BLAT через Интернет. Это более эффективно, поскольку позволяет хранить геном в памяти, в отличие от веб-приложения, которое хранит в памяти только индекс. [ 1 ] [ 9 ]
Лицензия
[ редактировать ]Как исходный код, так и предварительно скомпилированные двоичные файлы BLAT доступны бесплатно для академического и личного использования. Коммерческая лицензия на отдельный BLAT распространяется компанией Kent Informatics, Inc.
См. также
[ редактировать ]- BLAST Базовый инструмент поиска локального выравнивания
- Программное обеспечение для выравнивания последовательностей
Ссылки
[ редактировать ]- ^ а б с д и ж г час я дж к л м н Кент, В. Джеймс (2002). «BLAT — инструмент выравнивания, подобный BLAST» . Геномные исследования . 12 (4): 656–664. дои : 10.1101/гр.229202 . ПМК 187518 . ПМИД 11932250 .
- ^ а б с Имелфорт, Майкл (2009). Эдвардс, Д; Стаич, Дж; Хансен, Д. (ред.). Биоинформатика: инструменты и приложения . Нью-Йорк: Спрингер. стр. 19–20 . ISBN 978-0-387-92737-4 .
- ^ Нидлман, SB; Вунш, компакт-диск (1970). «Общий метод, применимый для поиска сходства в аминокислотной последовательности двух белков». Журнал молекулярной биологии . 48 (3): 443–53. дои : 10.1016/0022-2836(70)90057-4 . ПМИД 5420325 .
- ^ Смит, Т.Ф.; Уотерман, М.С. (1981). «Идентификация общих молекулярных подпоследовательностей». Журнал молекулярной биологии . 147 (1): 195–7. CiteSeerX 10.1.1.63.2897 . дои : 10.1016/0022-2836(81)90087-5 . ПМИД 7265238 .
- ^ Альтшул, Сан-Франциско; Гиш, В; Миллер, В; Майерс, EW; Липман, диджей (1990). «Базовый инструмент поиска локального выравнивания». Журнал молекулярной биологии . 215 (3): 403–10. дои : 10.1016/S0022-2836(05)80360-2 . ПМИД 2231712 . S2CID 14441902 .
- ^ Альтшул, Сан-Франциско; Мэдден, TL; Шеффер, А.А.; Чжан, Дж; Чжан, З; Миллер, В; Липман, диджей (1997). «Gapped BLAST и PSI-BLAST: новое поколение программ поиска по базам данных белков» . Исследования нуклеиновых кислот . 25 (17): 3389–402. дои : 10.1093/нар/25.17.3389 . ПМК 146917 . ПМИД 9254694 .
- ^ Баксеванис, Андреас Д.; Уэллетт, Б.Ф. Фрэнсис (2001). Биоинформатика: Практическое руководство по анализу генов и белков (2-е изд.). Нью-Йорк: Wiley-Interscience. стр. 187–214 . ISBN 978-0-471-22392-4 .
- ^ а б с д и ж г Сайт биоинформатики генома UCSC
- ^ а б с д и ж г час я дж к л м н Бхагват, Медха; Янг, Линн; Робисон, Рекс Р. (март 2012 г.). Использование BLAT для поиска сходства последовательностей в близкородственных геномах . 10.8. Том. 10. С. 10.8.1–10.8.24. дои : 10.1002/0471250953.bi1008s37 . ISBN 978-0-471-25095-1 . ПМК 4101998 . ПМИД 22389010 .
{{cite book}}:|journal=игнорируется ( помогите ) - ^ а б с д и Е, Шуй Цин (2008). Биоинформатика: практический подход . Лондон: Чепмен и Холл. стр. 11–12 . ISBN 978-1-58488-810-9 .
- ^ а б Кун, Р.М.; Хаусслер, Д; Кент, WJ (2013). «Браузер генома UCSC и связанные с ним инструменты» . Брифинги по биоинформатике . 14 (2): 144–61. дои : 10.1093/нагрудник/bbs038 . ПМК 3603215 . ПМИД 22908213 .
- ^ Лобо, Ингрид. «Базовый инструмент поиска локального выравнивания (BLAST)» . Природное образование . Проверено 15 октября 2013 г.
- ^ Певснер, Дж (2009). Биоинформатика и функциональная геномика . Нью-Джерси: John Wiley & Sons, Inc., стр. 166–167 . ISBN 978-0-470-08585-1 .
- ^ «NCBI – GenBank: AACZ03015565.1» . Проверено 12 октября 2013 г.