Опять секвенирование пептидов
В масс-спектрометрии секвенирование пептидов de novo представляет собой метод, при котором последовательность пептида аминокислотная определяется с помощью тандемной масс-спектрометрии .
Знание аминокислотной последовательности пептидов из гидролизата белка имеет важное значение для изучения биологической функции белка. Раньше это достигалось с помощью процедуры деградации Эдмана . [1] Сегодня анализ с помощью тандемного масс-спектрометра является более распространенным методом решения секвенирования пептидов. Обычно существует два подхода: поиск в базе данных и секвенирование de novo. Поиск в базе данных является простой версией, поскольку данные масс-спектров неизвестного пептида отправляются и обрабатываются для поиска соответствия с известной последовательностью пептида, будет выбран пептид с наивысшим показателем соответствия. [2] Этот подход не позволяет распознавать новые пептиды, поскольку он может сопоставляться только с существующими последовательностями в базе данных. Секвенирование de novo представляет собой выделение фрагментных ионов из масс-спектра. Различные алгоритмы [3] используются для интерпретации, и большинство инструментов поставляются с программами секвенирования de novo.
Фрагментация пептидов
[ редактировать ]Пептиды протонируются в режиме положительных ионов. Протон первоначально располагается на N-конце или на основном остатке боковой цепи, но из-за внутренней сольватации он может перемещаться вдоль основной цепи, разрываясь в разных местах, что приводит к образованию разных фрагментов. Правила фрагментации хорошо объяснены в некоторых публикациях. [4] [5] [6] [7] [8] [9]
Три различных типа связей основной цепи могут быть разорваны с образованием пептидных фрагментов: алкилкарбонильная (CHR-CO), пептидно-амидная связь (CO-NH) и аминоалкильная связь (NH-CHR). [ нужна ссылка ]
Различные типы фрагментных ионов
[ редактировать ]
При разрыве связей основной цепи образуются шесть различных типов ионов последовательности, как показано на рис. 1. Ионы N-концевого заряженного фрагмента классифицируются как a, b или c, а заряженные C-концевые ионы классифицируются как x, y. или з. Нижний индекс n обозначает количество аминокислотных остатков. Номенклатура была впервые предложена Репсторфом и Фольманом, затем Биман модифицировал ее, и эта версия стала наиболее широко распространенной. [11] [12]
Среди этих ионов последовательностей ионы a, b и y являются наиболее распространенными типами ионов, особенно в масс-спектрометрах низкоэнергетической диссоциации, индуцированной столкновениями (CID), поскольку пептид-амидная связь (CO-NH) является наиболее уязвимой и потеря CO из b-ионов.
Масса b-ионов = Σ (массы остатков) + 1 (H + )
Масса y-ионов = Σ (массы остатков) + 19 (H 2 O+H + )
Масса а-ионов = масса b-ионов – 28 (CO)
Двойное расщепление основной цепи приводит к образованию внутренних ионов ацильного типа, таких как H 2 N-CHR. 2 -CO-NH-CHR 3 -CO+ или иммониевого типа, например H 2 N-CHR 2 -CO-NH + =CHR 3 . Эти ионы обычно вносят помехи в спектры.
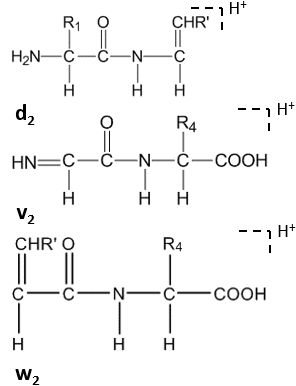
Дальнейшее расщепление происходит под действием высокоэнергетического CID по боковой цепи С-концевых остатков с образованием d n , v n , w n -ионов. [8]
Сводка правил фрагментации
[ редактировать ]Большинство фрагментных ионов представляют собой b- или y-ионы. a-ионы также часто наблюдаются при потере CO из b-ионов. [9]
Ионы-спутники (w n , v n , d n -ионы) образуются высокоэнергетическими CID.
Ионы, содержащие Ser-, Thr-, Asp- и Glu, вызывают нейтральную молекулярную потерю воды (-18).
Ионы, содержащие Asn-, Gln-, Lys-, Arg, вызывают нейтральные молекулярные потери аммиака (-17).
Нейтральная потеря аммиака из Arg приводит к образованию фрагментированных ионов (y-17) или (b-17) с более высоким содержанием, чем соответствующие им ионы.
Когда на С-конце имеется основной остаток, пептид генерирует ион (b n-1 +18).
Комплементарную по ионной паре можно наблюдать в спектрах многозарядных ионов. Для этого по ионной паре сумма их индексов равна общему числу аминокислотных остатков в неизвестном пептиде.
Если С-конец представляет собой Arg или Lys, y 1 в качестве доказательства можно найти в спектре ион .
Методы фрагментации пептидов
[ редактировать ]При низкоэнергетической диссоциации, индуцированной столкновениями (CID), b- и y-ионы являются основными ионами-продуктами. Кроме того, во фрагменте, содержащем аминокислоты RKNQ, наблюдается потеря аммиака (-17 Да). Потерю воды (-18 Да) можно наблюдать во фрагменте, содержащем в нем аминокислоты STED. Ионы-сателлиты в спектрах не показаны. [ нужна ссылка ]
В высокоэнергетическом CID можно наблюдать все различные типы фрагментированных ионов, но без потерь аммиака или воды. [ нужна ссылка ]
При диссоциации с переносом электрона (ETD) и диссоциации с захватом электрона (ECD) преобладающими ионами являются ионы c, y, z+1, z+2, а иногда и ионы w. [ нужна ссылка ]
Для распада после источника (PSD) в MALDI наиболее распространенными ионами-продуктами являются a, b, y-ионы. [ нужна ссылка ]
Факторами, влияющими на фрагментацию, являются зарядовое состояние (чем выше заряд, тем меньше энергии требуется для фрагментации), масса пептида (чем больше масса, тем больше энергии требуется), индуцированная энергия (более высокая энергия приводит к большей фрагментации), первичная аминокислотная последовательность, способ диссоциации и газ столкновения. [ нужна ссылка ]
Рекомендации по интерпретации
[ редактировать ]
Для интерпретации, [14] сначала найдите ионы аммония, состоящие из одной аминокислоты (H 2 N + =CHR 2 ). Соответствующие ионы иммония аминокислотам перечислены в таблице 1. Не обращайте внимания на несколько пиков на конце спектра с большой массой. Это ионы, которые теряют нейтральные молекулы (H 2 O, NH 3 , CO 2 , HCOOH) из [M+H] + ионы. Найдите разницу масс при 28 Да, поскольку b-ионы могут образовывать a-ионы за счет потери CO. Ищите b 2 -ионы на конце спектра с малой массой, что также помогает идентифицировать y n-2 -ионы. Массы b 2 -ионов указаны в таблице 2, а также отдельные аминокислоты, имеющие массу, равную b 2 -ионам. [15] Масса b 2 -иона = масса двух аминокислотных остатков + 1.

Определите серию ионов последовательности по той же разнице масс, которая соответствует массе одного из аминокислотных остатков (см. Таблицу 1). Например, различия масс между an и a n-1 , bn и bn -1 , c n и c n-1 одинаковы. Определите ион y n-1 на конце спектра с большой массой. Затем продолжайте идентифицировать ионы y n-2 , y n-3 ..., сопоставляя разницу масс с массами аминокислотных остатков (см. Таблицу 1). Найдите соответствующие b-ионы идентифицированных y-ионов. Масса ионов b+y равна массе пептида +2 Да. После определения серии y-иона и серии b-иона назначьте аминокислотную последовательность и проверьте массу. Другой метод состоит в том, чтобы сначала идентифицировать b-ионы, а затем найти соответствующие y-ионы. [ нужна ссылка ]
Алгоритмы и программное обеспечение
[ редактировать ]Ручное секвенирование de novo является трудоемким и требует много времени. Обычно алгоритмы или программы, поставляемые с масс-спектрометром, применяются для интерпретации спектров.
Разработка алгоритмов секвенирования de novo
[ редактировать ]Старый метод состоит в том, чтобы составить список всех возможных пептидов иона-предшественника в масс-спектре и сопоставить масс-спектр каждого кандидата с экспериментальным спектром. Возможный пептид, имеющий наиболее похожий спектр, будет иметь наибольшие шансы оказаться правильной последовательностью. Однако число возможных пептидов может быть большим. Например, пептид-предшественник с молекулярной массой 774 имеет 21 909 046 возможных пептидов. Несмотря на то, что это делается на компьютере, это занимает много времени. [17] [18]
Другой метод называется «субсеквенированием», при котором вместо перечисления всей последовательности возможных пептидов сопоставляются короткие последовательности пептидов, которые представляют собой только часть полного пептида. Когда обнаруживаются последовательности, которые полностью совпадают с ионами-фрагментами в экспериментальном спектре, они расширяются остатками один за другим, чтобы найти наилучшее соответствие. [19] [20] [21] [22]
В третьем методе применяется графическое отображение данных, при котором ионы-фрагменты, имеющие одинаковые различия масс одного аминокислотного остатка, соединяются линиями. Таким образом легче получить четкое изображение однотипных серий ионов. Этот метод может быть полезен для ручного секвенирования пептидов de novo, но не работает в условиях высокой производительности. [23]
Четвертый метод, который считается успешным, — это теория графов. Применение теории графов при секвенировании пептидов de novo было впервые упомянуто Бартельсом. [24] Пики спектра преобразуются в вершины графа, называемого «графом спектра». Если две вершины имеют одинаковую разницу масс одной или нескольких аминокислот, будет применено направленное ребро. Алгоритм SeqMS, [25] алгоритм Лютефиска, [26] Sherenga algorithm [27] Вот несколько примеров этого типа.
Глубокое обучение
[ редактировать ]Совсем недавно методы глубокого обучения были применены для решения проблемы секвенирования пептидов de novo. Первым прорывом стал DeepNovo, который принял структуру сверточной нейронной сети, добился значительного улучшения точности последовательности и позволил полностью собрать последовательность белка без помощи баз данных. [28] Впоследствии появились дополнительные сетевые структуры, такие как PointNet (PointNovo [29] ), были приняты для извлечения признаков из необработанного спектра. Проблема секвенирования пептидов de novo затем формулируется как проблема прогнозирования последовательности. Учитывая ранее предсказанную частичную последовательность пептида, модели секвенирования пептидов de novo на основе нейронных сетей будут неоднократно генерировать наиболее вероятную следующую аминокислоту до тех пор, пока масса предсказанного пептида не будет соответствовать массе предшественника. Во время вывода можно использовать стратегии поиска, такие как поиск по лучу, для исследования большего пространства поиска, сохраняя при этом низкие вычислительные затраты. По сравнению с предыдущими методами модели на основе нейронных сетей продемонстрировали значительно лучшую точность и чувствительность. [28] [29] [30] Более того, при тщательном проектировании модели алгоритмы секвенирования пептидов de novo на основе глубокого обучения также могут быть достаточно быстрыми для достижения секвенирования пептидов de novo в реальном времени. [29] Программное обеспечение PEAKS включает это обучение нейронной сети в свои алгоритмы секвенирования de novo.
Пакеты программного обеспечения
[ редактировать ]Как описано Андреотти и др. в 2012 году, [31] Антилопа представляет собой комбинацию лагранжевой релаксации и адаптации k кратчайших путей Йена. Он основан на методе «спектрального графика» и содержит различные оценочные функции, а по времени работы и точности может быть сопоставим с «популярными современными программами» PepNovo и NovoHMM.
Гроссманн и др. [32] представила AUDENS в 2005 году как автоматизированный инструмент для секвенирования пептидов de novo, содержащий модуль предварительной обработки, который может распознавать пики сигналов и пики шума.
Лютефиск может решить секвенирование de novo на основе масс-спектров CID. В этом алгоритме сначала обнаруживаются значимые ионы, а затем определяется список свидетельств N- и C-концевых. На основе списка последовательностей он генерирует полные последовательности в спектрах и сравнивает их с экспериментальным спектром. Однако результат может включать несколько кандидатов-последовательностей, которые имеют лишь небольшую разницу, поэтому трудно найти правильную последовательность пептида. Вторая программа, CIDentify, которая представляет собой модифицированную версию алгоритма FASTA Билла Пирсона, разработанную Алексом Тейлором, может применяться для различения неопределенных похожих кандидатов. [ нужна ссылка ]
Мо и др. представил алгоритм MSNovo в 2007 году и доказал, что он работает «лучше, чем существующие инструменты de novo, на нескольких наборах данных». [33] Этот алгоритм может выполнять de novo секвенирование масс-спектрометров LCQ, LTQ, а также одно-, двух- и трехзарядных ионов. В отличие от других алгоритмов, он применил новую оценочную функцию и использовал массив масс вместо графика спектра.
Фишер и др. [34] предложил метод секвенирования de novo NovoHMM. Скрытая модель Маркова (HMM) применяется как новый способ решения секвенирования de novo в байесовской системе. Вместо оценки отдельных символов последовательности этот метод учитывает апостериорные вероятности аминокислот. В статье на множестве примеров спектров доказано, что этот метод имеет лучшую производительность, чем другие популярные методы секвенирования пептидов de novo, такие как PepNovo.
PEAKS — это полный пакет программного обеспечения для интерпретации масс-спектров пептидов. Он включает в себя секвенирование de novo, поиск в базе данных, идентификацию PTM, поиск гомологии и количественный анализ данных. Ма и др. описал новую модель и алгоритм секвенирования de novo в PEAKS и сравнил эффективность с Лютефиском нескольких триптических пептидов стандартных белков с помощью квадрупольного времяпролетного масс-спектрометра (Q-TOF). [35]
PepNovo — это высокопроизводительный инструмент для секвенирования пептидов de novo, в котором в качестве метода оценки используется вероятностная сеть. Обычно интерпретация одного спектра занимает менее 0,2 секунды. Описано Франком и др. , PepNovo работает лучше, чем несколько популярных алгоритмов, таких как Sherenga, PEAKS, Lutefisk. [36] Теперь доступна новая версия PepNovo+.
Чи и др. представила pNovo+ в 2013 году как новый инструмент для секвенирования пептидов de novo с использованием комплементарных тандемных масс-спектров HCD и ETD. [37] В этом методе компонентный алгоритм pDAG значительно ускоряет время получения секвенирования пептидов в среднем до 0,018 с, что в три раза быстрее, чем в другом популярном программном обеспечении для секвенирования de novo.
Как описано Jeong et al. По сравнению с другими инструментами секвенирования пептидов do novo, которые хорошо работают только с определенными типами спектров, UniNovo является более универсальным инструментом, который имеет хорошую производительность на различных типах спектров или спектральных парах, таких как CID, ETD, HCD, CID/ETD, и т. д. Он имеет лучшую точность, чем PepNovo+ или PEAKS. Более того, это приводит к увеличению частоты ошибок обнаруженных пептидных последовательностей. [38]
Ма опубликовал Novor в 2015 году как машину для секвенирования пептидов de novo в реальном времени. Целью этого инструмента является повышение скорости de novo на порядок и сохранение такой же точности, как и у других инструментов de novo, представленных на рынке. На ноутбуке Macbook Pro Novor достиг более 300 спектров МС/МС в секунду. [39]
Певцов и др. сравнили производительность пяти вышеупомянутых алгоритмов секвенирования de novo: AUDENS, Lutefisk, NovoHMM, PepNovo и PEAKS. В анализе использовались данные масс-спектрометров QSTAR и LCQ, которые оценивались по значению относительного расстояния последовательности (RSD), которое представляло собой сходство между секвенированием пептида de novo и истинной последовательностью пептида, рассчитанной методом динамического программирования. Результаты показали, что все алгоритмы показали лучшую производительность в данных QSTAR, чем в данных LCQ, в то время как PEAKS как лучший имел показатель успеха 49,7% в данных QSTAR, а NovoHMM как лучший имел показатель успеха 18,3% в данных LCQ. Порядок производительности в данных QSTAR был PEAKS > Lutefisk, PepNovo > AUDENS, NovoHMM, а в данных LCQ - NovoHMM > PepNovo, PEAKS > Lutefisk > AUDENS. По сравнению с диапазоном качества спектра PEAKS и NovoHMM также показали лучшую производительность по обоим данным среди всех 5 алгоритмов. PEAKS и NovoHMM также имели лучшую чувствительность как по данным QSTAR, так и по данным LCQ. Однако ни один оцененный алгоритм не превысил 50% точности идентификации для обоих наборов данных. [40]
Последние достижения в области масс-спектрометров позволили генерировать масс-спектры сверхвысокого разрешения [1] . Повышенная точность вместе с увеличением объема генерируемых данных масс-спектрометрии вызывает интерес к применению методов глубокого обучения для секвенирования пептидов de novo. В 2017 году Тран и др. предложили DeepNovo, первое программное обеспечение для секвенирования de novo, основанное на глубоком обучении. Сравнительный анализ в оригинальной публикации показал, что DeepNovo значительно превзошёл предыдущие методы, включая PEAKS, Novor и PepNovo. DeepNovo реализован на Python с помощью платформы Tensorflow. [41] Чтобы представить массовый спектр как входные данные фиксированной размерности для нейронной сети, DeepNovo дискретизировала каждый спектр в вектор длиной 150 000. Это неоправданно большое представление спектра и однопоточная загрузка ЦП в исходной реализации не позволяют DeepNovo выполнять секвенирование пептидов в реальном времени. Для дальнейшего повышения эффективности моделей секвенирования пептидов de novo Qiao et al. предложил PointNovo в 2020 году. PointNovo — это программное обеспечение Python, реализованное с помощью платформы PyTorch. [42] и он избавляется от занимающего много места представления спектра-вектора, принятого DeepNovo. По сравнению с DeepNovo, PointNovo удалось одновременно достичь большей точности и эффективности за счет прямого представления спектра в виде набора пар m/z и интенсивности. [ нужна ссылка ]
Ссылки
[ редактировать ]- ^ Эдман, П.; Бегг, Г. (март 1967 г.). «Белковый секвенатор» . Европейский журнал биохимии . 1 (1): 80–91. дои : 10.1111/j.1432-1033.1967.tb00047.x . ПМИД 6059350 .
- ^ Уэбб-Робертсон, Б.-Дж.М.; Кэннон, WR (20 июня 2007 г.). «Современные тенденции в области вычислительных выводов на основе протеомики, основанной на масс-спектрометрии» . Брифинги по биоинформатике . 8 (5): 304–317. дои : 10.1093/нагрудник/bbm023 . ПМИД 17584764 .
- ^ Лу, Бингвэнь; Чен, Тин (март 2004 г.). «Алгоритмы секвенирования пептидов de novo с использованием тандемной масс-спектрометрии». Открытие лекарств сегодня: БИОСИЛИКО . 2 (2): 85–90. дои : 10.1016/S1741-8364(04)02387-X .
- ^ Jump up to: а б Папаяннопулос, Иоаннис А. (январь 1995 г.). «Интерпретация тандемных масс-спектров пептидов, вызванных столкновением». Обзоры масс-спектрометрии . 14 (1): 49–73. Бибкод : 1995MSRv...14...49P . дои : 10.1002/mas.1280140104 .
- ^ Дасс, Чхабил; Дезидерио, Доминик М. (май 1987 г.). «Масс-спектрометрический анализ опиоидных пептидов с помощью бомбардировки быстрыми атомами». Аналитическая биохимия . 163 (1): 52–66. дои : 10.1016/0003-2697(87)90092-3 . ПМИД 2887130 .
- ^ Ялчин, Талат; Чизмадиа, Имре Г.; Петерсон, Майкл Р.; Харрисон, Алекс Г. (март 1996 г.). «Структура и фрагментация ионов B n (n≥3) в пептидных спектрах» . Журнал Американского общества масс-спектрометрии . 7 (3): 233–242. дои : 10.1016/1044-0305(95)00677-X . ПМИД 24203294 .
- ^ Тан, Сюэ-Цзюнь; Бойд, Роберт К.; Бертран, MJ (ноябрь 1992 г.). «Исследование механизмов фрагментации дважды протонированных триптических пептидов». Быстрая связь в масс-спектрометрии . 6 (11): 651–657. Бибкод : 1992RCMS....6..651T . дои : 10.1002/rcm.1290061105 . ПМИД 1467549 .
- ^ Jump up to: а б с Джонсон, Ричард С.; Мартин, Стивен А.; Биманн, Клаус (декабрь 1988 г.). «Индуцированная столкновениями фрагментация ионов (M + H)+ пептидов. Ионы последовательности, специфичные для боковой цепи». Международный журнал масс-спектрометрии и ионных процессов . 86 : 137–154. Бибкод : 1988IJMSI..86..137J . дои : 10.1016/0168-1176(88)80060-0 .
- ^ Jump up to: а б Дасс, Чхабил (2007). Основы современной масс-спектрометрии ([Онлайн-Аусг.]. Под ред.). Хобокен, Нью-Джерси: Wiley-Interscience. стр. 317–322. дои : 10.1002/0470118490 . ISBN 9780470118498 .
- ^ Дасс, Чхабил (2001). Принципы и практика биологической масс-спектрометрии . Нью-Йорк, штат Нью-Йорк [ua]: Уайли. ISBN 978-0-471-33053-0 .
- ^ Ропсторф, П; Фолман, Дж. (ноябрь 1984 г.). «Предложение по общей номенклатуре ионов последовательности в масс-спектрах пептидов». Биомедицинская масс-спектрометрия . 11 (11): 601. doi : 10.1002/bms.1200111109 . ПМИД 6525415 .
- ^ Макклоски, Джеймс А., изд. (1990). Масс-спектрометрия . Сан-Диего: Академическая пресса. стр. 886–887. ISBN 978-0121820947 .
- ^ Фалик, AM; Хайнс, В.М.; Медзиградский, К.Ф.; Болдуин, Массачусетс; Гибсон, BW (ноябрь 1993 г.). «Ионы малой массы, полученные из пептидов путем высокоэнергетической диссоциации, вызванной столкновением, в тандемной масс-спектрометрии» . Журнал Американского общества масс-спектрометрии . 4 (11): 882–893. дои : 10.1016/1044-0305(93)87006-X . ПМИД 24227532 .
- ^ Дасс, Чхабил (2007). Основы современной масс-спектрометрии ([Онлайн-Аусг.]. Под ред.). Хобокен, Нью-Джерси: Wiley-Interscience. стр. 327–330. ISBN 9780470118498 .
- ^ Харрисон, Алекс Г.; Чизмадиа, Имре Г.; Тан, Тин-Хуа (май 2000 г.). «Структура и фрагментация ионов b 2 в масс-спектрах пептидов». Журнал Американского общества масс-спектрометрии . 11 (5): 427–436. дои : 10.1016/S1044-0305(00)00104-5 . ПМИД 10790847 . S2CID 24794690 .
- ^ Дасс, Чхабил (2007). Основы современной масс-спектрометрии ([Онлайн-Аусг.]. Под ред.). Хобокен, Нью-Джерси: Wiley-Interscience. стр. 329. ИСБН 9780470118498 .
- ^ Сакурай, Т.; Мацуо, Т.; Мацуда, Х.; Катакусе, И. (август 1984 г.). «PAAS 3: Компьютерная программа для определения вероятной последовательности пептидов на основе масс-спектрометрических данных». Биологическая масс-спектрометрия . 11 (8): 396–399. дои : 10.1002/bms.1200110806 .
- ^ Хамм, CW; Уилсон, МЫ; Харван, диджей (1986). «Программа секвенирования пептидов». Биоинформатика . 2 (2): 115–118. дои : 10.1093/биоинформатика/2.2.115 . ПМИД 3450361 .
- ^ Биманн, К; Конус, С; Вебстер, Британская Колумбия; Арсено, врач общей практики (5 декабря 1966 г.). «Определение аминокислотной последовательности в олигопептидах путем компьютерной интерпретации их масс-спектров высокого разрешения». Журнал Американского химического общества . 88 (23): 5598–606. дои : 10.1021/ja00975a045 . ПМИД 5980176 .
- ^ Исикава, К.; Нива, Ю. (июль 1986 г.). «Компьютерное секвенирование пептидов методом масс-спектрометрии с бомбардировкой быстрыми атомами». Биологическая масс-спектрометрия . 13 (7): 373–380. дои : 10.1002/bms.1200130709 .
- ^ Сигел, ММ; Бауман, Н. (15 марта 1988 г.). «Эффективный алгоритм секвенирования пептидов с использованием данных масс-спектральной бомбардировки быстрыми атомами». Биомедицинская и экологическая масс-спектрометрия . 15 (6): 333–43. дои : 10.1002/bms.1200150606 . ПМИД 2967723 .
- ^ Джонсон, РС; Биманн, К. (ноябрь 1989 г.). «Компьютерная программа (SEQPEP) для помощи в интерпретации масс-спектров тандемных столкновений пептидов при высоких энергиях». Биомедицинская и экологическая масс-спектрометрия . 18 (11): 945–57. дои : 10.1002/bms.1200181102 . ПМИД 2620156 .
- ^ Скобл, Хьюберт А.; Биллер, Джеймс Э.; Биманн, Клаус (1987). «Стратегия, ориентированная на графическое отображение, для секвенирования аминокислот пептидов с помощью тандемной масс-спектрометрии». Журнал Фрезениуса по аналитической химии . 327 (2): 239–245. дои : 10.1007/BF00469824 . S2CID 97665981 .
- ^ Бартельс, Кристиан (июнь 1990 г.). «Быстрый алгоритм секвенирования пептидов методом масс-спектроскопии». Биологическая масс-спектрометрия . 19 (6): 363–368. дои : 10.1002/bms.1200190607 . ПМИД 24730078 .
- ^ Фернандес-де-Коссио, Дж; Гонсалес, Дж; Бесада, В. (август 1995 г.). «Компьютерная программа, помогающая секвенировать пептиды в экспериментах по разложению, активируемому столкновением». Компьютерные приложения в биологических науках . 11 (4): 427–34. дои : 10.1093/биоинформатика/11.4.427 . ПМИД 8521052 .
- ^ Тейлор, Дж.А.; Джонсон, Р.С. (1997). «Поиск в базе данных последовательностей с помощью секвенирования пептидов de novo с помощью тандемной масс-спектрометрии». Быстрая связь в масс-спектрометрии . 11 (9): 1067–75. Бибкод : 1997RCMS...11.1067T . doi : 10.1002/(sici)1097-0231(19970615)11:9<1067::aid-rcm953>3.0.co;2-l . ПМИД 9204580 .
- ^ Данчик, Владо; Аддона, Тереза А.; Клаузер, Карл Р.; Ват, Джеймс Э.; Певзнер, Павел А. (октябрь 1999 г.). «Секвенирование пептидов с помощью тандемной масс-спектрометрии». Журнал вычислительной биологии . 6 (3–4): 327–342. CiteSeerX 10.1.1.128.2645 . дои : 10.1089/106652799318300 . ПМИД 10582570 .
- ^ Jump up to: а б Тран, Нгок Хиеу и др. «Секвенирование пептидов de novo методом глубокого обучения». Труды Национальной академии наук 114.31 (2017): 8247-8252.
- ^ Jump up to: а б с Цяо, Руи и др. «Независимое от вычислительного разрешения секвенирование пептидов de novo для устройств с высоким разрешением». Природный машинный интеллект 3.5 (2021 г.): 420-425.
- ^ Карунратанакул, Корраве и др. «Открытие тысяч новых пептидов с помощью гибридной структуры секвенирования пептидов de novo с помощью поиска последовательности-маски». Молекулярная и клеточная протеомика 18.12 (2019): 2478-2491.
- ^ Андреотти, С; Клау, ГВ; Райнерт, К. (2012). «Антилопа - подход лагранжевой релаксации к проблеме секвенирования пептидов de novo». Транзакции IEEE/ACM по вычислительной биологии и биоинформатике . 9 (2): 385–94. arXiv : 1102.4016 . дои : 10.1109/tcbb.2011.59 . ПМИД 21464512 . S2CID 593303 .
- ^ Гроссманн, Дж; Роос, ФФ; Челебак, М; Липтак, З; Матис, ЛК; Мюллер, М; Грюиссем, В; Багинский, С (2005). «AUDENS: инструмент для автоматического секвенирования пептидов de novo». Журнал исследований протеома . 4 (5): 1768–74. CiteSeerX 10.1.1.654.169 . дои : 10.1021/pr050070a . ПМИД 16212431 .
- ^ Мо, Л; Дутта, Д; Ван, Ю; Чен, Т. (1 июля 2007 г.). «MSNovo: алгоритм динамического программирования для секвенирования пептидов de novo с помощью тандемной масс-спектрометрии». Аналитическая химия . 79 (13): 4870–8. дои : 10.1021/ac070039n . ПМИД 17550227 .
- ^ Фишер, Б; Рот, В; Роос, Ф; Гроссманн, Дж; Багинский С; Видмайер, П; Грюиссем, В; Буманн, Дж. М. (15 ноября 2005 г.). «NovoHMM: скрытая марковская модель для секвенирования пептидов de novo». Аналитическая химия . 77 (22): 7265–73. CiteSeerX 10.1.1.507.1610 . дои : 10.1021/ac0508853 . ПМИД 16285674 .
- ^ Ма, Бин; Чжан, Кайчжун; Хендри, Кристофер; Лян, Чэнчжи; Ли, Мин; Доэрти-Кирби, Аманда; Лажуа, Жиль (30 октября 2003 г.). «PEAKS: мощное программное обеспечение для секвенирования пептидов de novo методом тандемной масс-спектрометрии». Быстрая связь в масс-спектрометрии . 17 (20): 2337–2342. Бибкод : 2003RCMS...17.2337M . дои : 10.1002/rcm.1196 . ПМИД 14558135 .
- ^ Франк, А; Певзнер, П. (15 февраля 2005 г.). «PepNovo: секвенирование пептидов de novo посредством вероятностного сетевого моделирования». Аналитическая химия . 77 (4): 964–73. дои : 10.1021/ac048788h . ПМИД 15858974 .
- ^ Чи, Х; Чен, Х; Он, К; Ву, Л; Ян, Б; Солнце, RX; Лю, Дж; Цзэн, ВФ; Сонг, CQ; Он, С.М.; Донг, MQ (1 февраля 2013 г.). «pNovo+: секвенирование пептидов de novo с использованием комплементарных тандемных масс-спектров HCD и ETD». Журнал исследований протеома . 12 (2): 615–25. дои : 10.1021/pr3006843 . ПМИД 23272783 .
- ^ Чон, К; Ким, С; Певзнер, Пенсильвания (15 августа 2013 г.). «UniNovo: универсальный инструмент для секвенирования пептидов de novo» . Биоинформатика . 29 (16): 1953–62. doi : 10.1093/биоинформатика/btt338 . ПМЦ 3722526 . ПМИД 23766417 .
- ^ Ма, Бин (30 июня 2015 г.). «Novor: программное обеспечение для секвенирования пептидов de Novo в реальном времени» . Журнал Американского общества масс-спектрометрии . 26 (11): 1885–1894. Бибкод : 2015JASMS..26.1885M . дои : 10.1007/s13361-015-1204-0 . ПМК 4604512 . ПМИД 26122521 .
- ^ Певцов С.; Федулова И.; Мирзаи, Х.; Бак, К.; Чжан, X. (2006). «Оценка производительности существующих De Novo алгоритмов секвенирования ». Журнал исследований протеома . 5 (11): 3018–3028. дои : 10.1021/pr060222h . ПМИД 17081053 .
- ^ Абади, Мартин и др. «Tensorflow: система крупномасштабного машинного обучения». 12-й симпозиум {USENIX} по проектированию и внедрению операционных систем ({OSDI} 16). 2016.
- ^ Адам и др. «Pytorch: высокопроизводительная библиотека глубокого обучения в императивном стиле». Достижения в области нейронных систем обработки информации 32 (2019): 8026-8037.