Реакция Стеттера
| Реакция Стеттера | |
|---|---|
| Назван в честь | Герман Стеттер |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | Стеттер-реакция |
Реакция Стеттера — это реакция, используемая в органической химии для образования углерод-углеродных связей посредством реакции 1,4-присоединения с использованием нуклеофильного катализатора . [1] Хотя родственная реакция 1,2-присоединения , бензоиновая конденсация , была известна с 1830-х годов, о реакции Стеттера не сообщалось до 1973 года доктором Германом Стеттером. [2] Реакция дает синтетически полезные 1,4-дикарбонильные соединения и родственные производные альдегидов и акцепторов Михаэля . В отличие от 1,3-дикарбонилов, которые легко получить посредством конденсации Кляйзена , или 1,5-дикарбонилов, которые обычно получают с помощью реакции Михаэля , 1,4-дикарбонилы являются сложными для синтеза субстратами, но являются ценными исходными материалами для нескольких , в том числе синтез Паала–Кнорра фуранов органические превращения и пирролов. Традиционно используемыми катализаторами реакции Стеттера являются соли тиазолия и цианид-анион, но более поздние работы по асимметричной реакции Стеттера показали, что соли триазолия эффективны. Реакция Стеттера является примером химии умполунга , поскольку присущая альдегиду полярность меняется на противоположную при добавлении катализатора к альдегиду, что делает углеродный центр нуклеофильным, а не электрофильным.

Механизм
[ редактировать ]Поскольку реакция Стеттера является примером химии умполунга , альдегид превращается из электрофила в нуклеофил в условиях реакции. [3] Это достигается путем активации какого-либо катализатора - либо цианида (CN − ) или тиазолиевую соль. [1] Механизм использования любого катализатора очень похож; единственное отличие состоит в том, что в случае с солями тиазолия катализатор необходимо сначала депротонировать для образования активных каталитических частиц. Активный катализатор можно описать как комбинацию двух способствующих резонансу форм — илида или карбена , обе из которых характеризуются нуклеофильным характером углерода. Илид тиазолия или CN − затем можно добавить к альдегидному субстрату, образуя циангидрин в случае CN. − или промежуточное соединение Бреслоу в случае соли тиазолия. Промежуточное соединение Бреслоу было предложено Рональдом Бреслоу в 1958 году и является общим промежуточным соединением для всех тиамином реакций, катализируемых , как in vitro , так и in vivo . [4]

«нуклеофильного альдегида» После образования синтона , будь то в виде циангидрина или стабилизированного тиазолийилидом, реакция может идти по двум путям. Более быстрый путь — самоконденсация с другой молекулой альдегида с образованием бензоиновых продуктов. Однако конденсация бензоина полностью обратима и поэтому не препятствует образованию продуктов реакции Стеттера. Фактически, бензоины можно использовать вместо альдегидов в качестве субстратов для достижения того же общего преобразования Стеттера, поскольку бензоины могут быть восстановлены до своих альдегидных предшественников в условиях реакции. [1] Желаемый путь к продукту Стеттера - это 1,4-присоединение нуклеофильного альдегида к акцептору типа Михаэля. После 1,4-присоединения реакция необратима, и в конечном итоге 1,4-дикарбонил образуется при удалении катализатора для регенерации CN. − или тиазолий илид.

Объем
[ редактировать ]Реакция Стеттера дает классически труднодоступные 1,4-дикарбонильные соединения и родственные производные. Традиционная реакция Стеттера весьма универсальна и работает на самых разных субстратах. [1] Ароматические альдегиды, гетероароматические альдегиды и бензоины могут быть использованы в качестве предшественников ациланионов с тиазолиевой солью и цианидными катализаторами. Однако алифатические альдегиды можно использовать только в том случае, если в качестве катализатора используется соль тиазолия, поскольку при использовании цианидного катализатора они подвергаются побочной реакции альдольной конденсации. Кроме того, α,β-ненасыщенные сложные эфиры, кетоны, нитрилы, нитросы и альдегиды являются подходящими акцепторами Михаэля для любого катализатора. Однако общий объем асимметричных реакций Стеттера более ограничен. Внутримолекулярные асимметричные реакции Стеттера используют ряд приемлемых акцепторов Михаэля и предшественников ациланионов практически в любой комбинации. [5] Внутримолекулярные асимметричные реакции Стеттера могут использовать ароматические, гетероароматические и алифатические альдегиды со связанным α,β-ненасыщенным сложным эфиром, кетоном, тиоэфиром, малонатом, нитрилом или амидом Вейнреба. Было показано, что α,β-ненасыщенные нитро и альдегиды не являются подходящими акцепторами Михаэля и заметно уменьшают энантиомерный избыток в таких реакциях. [5] Другое ограничение, с которым сталкиваются внутримолекулярные асимметричные реакции Стеттера, заключается в том, что только субстраты, которые приводят к образованию шестичленного кольца, демонстрируют синтетически полезный энантиомерный избыток; субстраты, образующие пяти- и семичленные кольца, либо не реагируют, либо обладают низкой стереоиндукцией. [5] С другой стороны, межмолекулярные асимметричные реакции в значительной степени ограничиваются специально подобранными комбинациями предшественника ациланиона и акцептора Михаэля, таких как алифатический альдегид с нитроалкеном. [6] Кроме того, эти субстраты склонны к активации, поскольку межмолекулярная асимметричная реакция Стеттера все еще находится на ранних стадиях развития.

Вариации
[ редактировать ]С момента ее открытия в 1973 году было разработано несколько вариантов реакции Стеттера. В 2001 году Марри и др. сообщили о реакции Стеттера ароматических альдегидов на производные ациламина с образованием продуктов α-амидокетонов. [7] Акцепторы ацилимина генерировались in situ из субстратов α-тозиламида, подвергавшихся элиминации в присутствии основания. Наблюдались выходы от хороших до отличных (75-90%). Механистические исследования показали, что соответствующие бензоины не являются адекватными субстратами, в отличие от традиционных реакций Стеттера. [1] Из этого авторы делают вывод, что реакция Стеттера ацилиминов находится под кинетическим, а не термодинамическим контролем.

Другой вариант реакции Стеттера включает использование 1,2-дикарбонилов в качестве предшественников промежуточного ациланиона. В 2005 году Шейдт и его коллеги сообщили об использовании пирувата натрия, который теряет CO 2 с образованием промежуточного продукта Бреслоу. [8] Аналогичным образом, в 2011 году Бортолини и его коллеги продемонстрировали использование α-дикетонов для генерации ацил-аниона. [9] В разработанных ими условиях 2,3-бутадиенон расщепляется после добавления к тиазолиевому катализатору с высвобождением этилацетата и образованием промежуточного продукта Бреслоу, необходимого для протекания реакции Стеттера.

Кроме того, они показали экономию атомов и полезность использования циклического α-дикетона для получения продукта Стеттера со связанным этиловым эфиром. Реакция протекает по тому же механизму, что и ациклическая версия, но сложный эфир, образующийся при воздействии этанола, остается связанным с продуктом. Однако условия позволяют получать только этиловые эфиры из-за необходимости использования этанола в качестве растворителя. Замена этанола трет -бутанолом не привела к получению продукта. Авторы предполагают, что это связано с разницей в кислотности двух спиртовых растворителей.
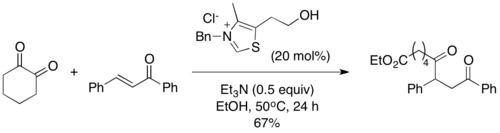
В 2004 году Шейдт и его коллеги представили ацилсиланы в качестве компетентных субстратов в реакции Стеттера, вариант, который они назвали «реакцией сила-Стеттер». [10] В условиях реакции тиазолиевый катализатор вызывает перегруппировку [1,2] Брука , за которой следует десилилирование добавкой изопропанола с образованием обычного промежуточного продукта Бреслоу традиционной реакции Стеттера. Установлено, что стадия десилилирования необходима, и реакция не протекает без спиртовой добавки. Ацилсиланы менее электрофильны, чем соответствующие альдегиды, что предотвращает образование типичных побочных продуктов бензоинового типа, часто наблюдаемых в реакции Стеттера. [11]
Асимметричная реакция Стеттера
[ редактировать ]О первом асимметричном варианте реакции Стеттера сообщили в 1996 году Эндерс и др . с использованием хирального триазолиевого катализатора 1 . [12] Впоследствии сообщалось о нескольких других катализаторах асимметричных реакций Стеттера, в том числе 2 , [13] 3 , [14] и 4 . [15]

Успех катализатора 2 группы Rovis побудил их к дальнейшему исследованию этого семейства катализаторов и расширению их использования для асимметричных реакций Стеттера. В 2004 году они сообщили об энантиоселективном образовании четвертичных центров из ароматических альдегидов во внутримолекулярной реакции Стеттера со слегка модифицированным катализатором. [16] Дальнейшие работы расширили сферу действия этой реакции, включив в нее также алифатические альдегиды. [17] Впоследствии было показано, что олефиновая геометрия акцептора Михаэля диктует диастереоселективность в этих реакциях, при этом катализатор диктует энантиоселективность образования начальной углеродной связи, а минимизация аллильной деформации диктует диастереоселективное внутримолекулярное протонирование. [18]

Неизбежные трудности контроля энантиоселективности в межмолекулярных реакциях сделали разработку межмолекулярной асимметричной реакции Стеттера сложной задачей. Хотя в начале 1990-х годов Эндерс сообщил об ограниченном энантиомерном избытке реакции н -бутаналя с халконом, [19] об условиях для синтетически полезной асимметричной межмолекулярной реакции Стеттера не сообщалось до 2008 года, когда обе группы Эндерса и Ровиса опубликовали такие реакции. Группа Эндерса использовала катализатор на основе триазолия для проведения реакции сочетания ароматических альдегидов с производными халкона с умеренными выходами. [20] В параллельной публикации группы Ровиса также использовался катализатор на основе триазолия и сообщалось о реакции Стеттера между глиоксамидами и алкилиденмалонатами с хорошими и отличными выходами. [21]

Ровис и его коллеги впоследствии продолжили исследование асимметричной межмолекулярной реакции Стеттера гетероциклических альдегидов и нитроалкенов . [22] В ходе оптимизации этой реакции было обнаружено, что катализатор с фторированной основной цепью значительно повышает энантиоселективность реакции. Было высказано предположение, что фторированная основная цепь помогает зафиксировать конформацию катализатора таким образом, что повышается энантиоселективность. Дальнейшие вычислительные исследования этой системы подтвердили, что стереоэлектронное притяжение между развивающимся частичным отрицательным зарядом нитроалкена в переходном состоянии и частичным положительным зарядом диполя CF ответственно за увеличение энантиомерного избытка, наблюдаемое при использовании катализатора с основной цепью. фторирование. [23] Хотя это значительный прогресс в области межмолекулярных асимметричных реакций Стеттера, объем субстратов ограничен, а катализатор оптимизирован для конкретных используемых субстратов.

Еще один вклад в развитие асимметричных межмолекулярных реакций Стеттера был сделан Глориусом и его коллегами в 2011 году. [6] Они продемонстрировали энантиоселективный синтез α-аминокислот, используя N -ациламидоакрилат в качестве акцептора конъюгата. Примечательно, что реакцию можно проводить в масштабе 5 ммоль без потери выхода или энантиоселективности.

Приложения
[ редактировать ]Реакция Стеттера — эффективный инструмент органического синтеза . Продукты реакции Стеттера — 1,4-дикарбонилы — являются ценными фрагментами для синтеза сложных молекул. Например, Трост и его коллеги использовали реакцию Стеттера в качестве одного из этапов синтеза рац -гирсутиновой кислоты C. [24] Внутримолекулярное сочетание алифатического альдегида со связанным α,β-ненасыщенным эфиром привело к получению желаемого трициклического 1,4-дикарбонила с выходом 67%. Это промежуточное соединение было превращено в рац -гирсутиновую кислоту C еще за семь стадий.

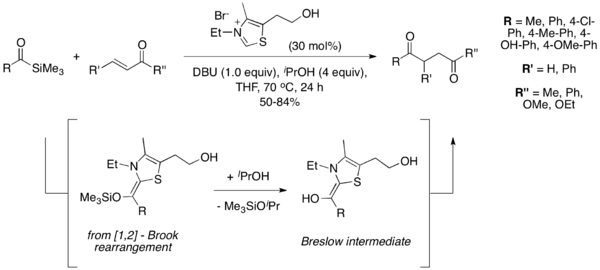
Реакция Стеттера обычно используется последовательно с синтезом фуранов и пирролов Паала-Кнорра , при котором 1,4-дикарбонил подвергается конденсации сам с собой или в присутствии амина при высокой температуре и кислых условиях. В 2001 году Тиус и его коллеги сообщили об асимметричном полном синтезе розеофилина с использованием межмолекулярной реакции Стеттера для соединения алифатического альдегида с циклическим еноном. [25] После метатезиса с замыканием цикла и восстановления алкена 1,4-дикарбонильный продукт был превращен в пиррол посредством синтеза Паала-Кнорра и далее преобразован в природный продукт.
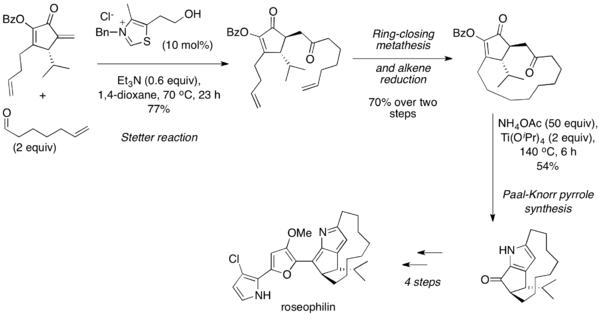
В 2004 году было опубликовано сообщение о последовательности сочетания-изомеризации-Стеттера-Паала Кнорра в одном горшке. [26] В этой процедуре сначала используется химия перекрестного сочетания палладия для соединения арилгалогенидов с пропаргиловыми спиртами с образованием α,β-ненасыщенных кетонов, которые затем могут подвергаться реакции Стеттера с альдегидом. После образования 1,4-дикарбонильного соединения нагревание в присутствии кислоты дает фуран, а нагревание в присутствии хлорида аммония и кислоты дает пиррол. Вся последовательность выполняется в одном сосуде без обработки или очистки между этапами.

Ма и его коллеги разработали альтернативный метод получения фуранов с использованием реакции Стеттера. [27] В их отчете 3-аминофураны синтезированы в условиях Стеттера для сочетания ароматических альдегидов с диметилацетилендикарбоксилатом (ДМАД), при этом тиазолийилид гидролизуется путем ароматизации фуранового продукта. Поскольку тиазолий в этих условиях разрушается, он не является каталитиком и его следует использовать в стехиометрических количествах.
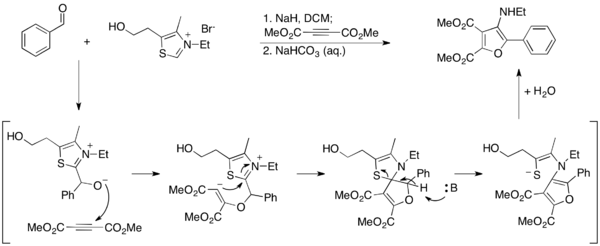
Они продолжили эту работу, разработав метод, в котором 2-аминофураны синтезируются путем циклизации на нитрил. [28] В этом методе тиазолийилид используется в качестве катализатора и образуется свободный аминный продукт.

Связанный
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с д и Стеттер, Х. Энджью. хим. Межд. Эд. 1976 , 15 , 639.
- ^ Стеттер, Х. и Шокенберг, М. Ангью. хим. Эд. англ. 1973 , 12 , 81.
- ^ Олбрайт, Дж. Д. Тетраэдр 1983 , 39 , 3207.
- ^ Бреслоу, RJ Am. хим. Соц. 1958 , 80 , 3719.
- ^ Jump up to: а б с де Аланис-младший; Керр, MS; Мур, Дж.Л.; Ровис, TJ Org. хим. 2008 , 73 , 2033.
- ^ Jump up to: а б Жуссом, Т.; Вурц, штат Невада; Глориус, Ф. Энджью. хим. Межд. Эд. 2011 , 50 , 1410.
- ^ Марри, Дж.А.; Франц, Делавэр; Сохейли, А.; Тиллер, Р.; Грабовский, EJJ; Рейдер, PJ J. Am. хим. Соц. 2001 , 123 , 9696.
- ^ Майерс, MC; Бхарадвадж, Арканзас; Милгрэм, Британская Колумбия; Шайдт, К.А. Дж. Ам. хим. Соц. 2005 , 127 , 14675.
- ^ Бортолини, О.; Фантен, Г.; Фоганьоло, М.; Джон, ПП; Масси, А.; Пасифико, С. Орг. Биомол. хим. 2011 , 9 , 8437.
- ^ Мэттсон, AE; Бхарадвадж, Арканзас; Шайдт, К.А. Дж. Ам. хим. Соц. 2004 , 126 , 2314.
- ^ Мэттсон, AE; Бхарадвадж, Арканзас; Зуль, AM; Шайдт, К.А. «Тиазолий-катализируемые присоединения ацилсиланов: общая стратегия реакций присоединения ацил-анионов». Дж. Орг. хим. 2006 , 71 , 5715. два : 10.1021/jo060699c
- ^ Эндерс, Д.; Брат К.; Рансинк, Дж.; Телес, Дж. Х. Хелв. Сказать. Акта 1996 , 79 , 1899.
- ^ Керр, MS; де Аланис-младший; Ровис, T.J. Am. хим. Соц. 2002 , 124 , 10298.
- ^ Пеш, Дж.; Хармс, К.; Бах, T. Eur. Chem. 2004 , 2025.
- ^ Меннен, С.М.; Бланк, Джей Ти; Тран-Дюбе, МБ; Имбриглио, Дж. Э.; Миллер, SJ Chem. Общий. 2005 , 195.
- ^ Керр, MS; Ровис, T.J. Am. хим. Соц. 2004 , 126 , 8876.
- ^ Мур, Дж.Л.; Керр, MS; Ровис, Т. Тетраэдр 2006 , 62 , 11477.
- ^ де Аланис, младший; Ровис, T.J. Am. хим. Соц. 2005 , 127 , 6284.
- ^ Эндерс, Д. Образование ферментемиметических связей CC и CN. В стереоселективном синтезе ; Оттоу Э., Шелькопф К., Шульц Б.-Г., ред.; Springer-Verlag: Берлин-Гейдельберг, 1994; стр. 63-90.
- ^ Эндерс, Д.; Хан, Дж.; Хенселер, А. Хим. Общий. 2008 , 3989.
- ^ Лю, К.; Перро, С.; Ровис, T.J. Am. хим. Соц. 2008 , 130 , 14066.
- ^ ДиРокко, Д.А.; Оберг, К.М.; Далтон, DM; Ровис, T.J. Am. хим. Соц. 2009 , 131 , 10872.
- ^ Хм, Дж. М.; ДиРокко, Д.А.; Ной, Эл; Ровис, Т.; Хоук, KN J. Am. хим. Соц. 2011 , 133 , 11249.
- ^ Трост, Б.М.; Шуи, компакт-диск; ДиНинно Ф.-младший; МакЭлвейн, SS J. Am. хим. Соц. 1979 , 101 , 1284.
- ^ Харрингтон, ЧП; Тиус, Массачусетс Дж. Ам. хим. Соц. 2001 , 123 , 8509.
- ^ Браун, РУ; Мюллер, TJJ Synthesis 2004 , 14 , 2391.
- ^ Ма, К.; Ян, Y. Org Lett. 2005 , 7 , 1343.
- ^ Лю, П.; Лей, М.; Ма, Л.; Ху, Л. Синлетт 2011 , 8 , 1133.