Принцип Кертина – Хэммета
Принцип Кертина-Хэммета — это принцип химической кинетики, предложенный Дэвидом Ярроу Кертином и Луи Плаком Хэмметом . В нем говорится, что для реакции, в которой есть пара реакционноспособных промежуточных продуктов или реагентов , которые быстро взаимопревращаются (как это обычно бывает с конформационными изомерами ), каждый из которых необратимо переходит в другой продукт, соотношение продуктов будет зависеть как от разницы в энергии между два конформера и энергетические барьеры от каждого из быстро уравновешивающих изомеров к их соответствующим продуктам. Другими словами, распределение продуктов отражает разницу в энергии между двумя переходными состояниями, ограничивающими скорость. В результате распределение продуктов не обязательно будет отражать равновесное распределение двух промежуточных продуктов. [1] [2] Принцип Кертина-Хаммета был использован для объяснения селективности во множестве стерео- и региоселективных реакций. Связь между (кажущимися) константами скорости и константой равновесия известна как уравнение Уинстейна - Холнесса .
Определение [ править ]
Принцип Кертина-Хаммета применим к системам, в которых разные продукты образуются из двух субстратов, находящихся в равновесии друг с другом. Быстро взаимопревращающиеся реагенты могут иметь между собой любые взаимоотношения ( стереоизомеры , конституциональные изомеры , конформационные изомеры и т. д.). Формирование продуктов должно быть необратимым, и различные продукты не должны подвергаться взаимному преобразованию. [3]
Например, даны виды A и B , которые быстро уравновешиваются, в то время как A необратимо превращается в C , а B необратимо превращается в D :
![{\displaystyle {\ce {\bf {{C}\ {\it {<-[k_{\rm {1}}]{\bf {{A}{\it {\ <=>[{K}] \ {\bf {{B}\ {\it {->[k_{\rm {2}}]\ {\bf {D}}}}}}}}}}}}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8bf279b211853c9be67aece08ca6504276d4b50d)
K — константа равновесия между A и B , а k 1 и k 2 — константы скорости образования C и D соответственно. Когда скорость взаимного превращения между A и B намного выше, чем k 1 или k 2 , тогда принцип Кертина-Хаммета говорит нам, что соотношение продуктов C : D не равно равновесному соотношению реагентов A : B , а вместо этого определяется относительными энергиями переходных состояний (т. е. разностью абсолютных энергий переходных состояний). Если бы реагенты A и B имели одинаковую энергию, соотношение продуктов зависело бы только от активационных барьеров реакций, приводящих к каждому соответствующему продукту. Однако в реальном сценарии два реагента, вероятно, находятся на несколько разных энергетических уровнях, хотя барьер для их взаимного превращения должен быть низким, чтобы можно было применить сценарий Кертина-Хэммета. В этом случае распределение продуктов зависит как от равновесного соотношения А и В, так и от относительных активационных барьеров, идущих к соответствующим продуктам. С и Д. Оба фактора учитываются разницей энергий переходных состояний (ΔΔ G ‡ на рисунке ниже).
Профиль свободной энергии координат реакции типичной реакции под контролем Кертина-Хаммета представлен следующим рисунком:
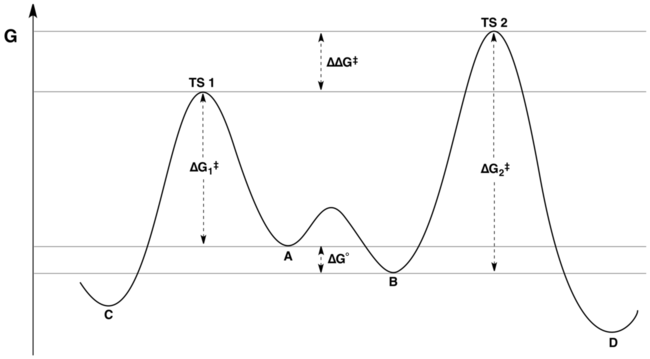
Соотношение продуктов зависит только от значения, обозначенного ΔΔ G ‡ на рисунке: C будет основным продуктом, поскольку энергия TS1 ниже энергии TS2 . Распространенным, но ложным утверждением является то, что распределение продуктов никоим образом не отражает относительную свободную энергию субстратов A и B ; фактически, он отражает относительные свободные энергии субстратов и относительные энергии активации. [3] [4] Это недоразумение может возникнуть из-за непонимания различия между «разницей энергий активации» и «разницей в энергиях переходного состояния». Хотя эти величины на первый взгляд могут показаться синонимами, последняя учитывает константу равновесия для взаимного превращения A и B , а первая - нет.
Математически соотношение продуктов можно выразить как функцию K , k 1 и k 2 или через соответствующие энергии Δ G °, Δ G 1 ‡ , и Δ G 2 ‡ . Комбинируя слагаемые, соотношение продуктов можно переписать через величину ΔΔ G ‡ в одиночку, где ΔΔ G ‡ = (Δ G 2 ‡ – Δ Г 1 ‡ ) + ΔG ° . Анализ энергетической диаграммы (показанной выше) показывает, что ΔΔ G ‡ как раз и заключается разница в энергиях переходных состояний.
Вывод [ править ]
Типовую реакцию Кертина – Хэммета можно описать следующими параметрами:
Чтобы быстрое уравновешивание было хорошим предположением, скорость превращения менее стабильного продукта A или B в продукт C или D чем скорость установления равновесия между A и B. должна быть как минимум в 10 раз медленнее , [5]
Скорость образования соединения C из A определяется как
- ,
![{\displaystyle {\frac {d[\mathbf {C} ]}{dt}}=k_ {1}[\mathbf {A} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7a0ba64b7c5b396fc651fb3aad19d80574dcdcdc)
и D из B как
- ,
![{\displaystyle {\frac {d[\mathbf {D} ]}{dt}}=k_ {2}[\mathbf {B} ]\approx k_{2}K[\mathbf {A} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e28151c0c25112e0df3d966f7dd520e4e3071c7f)
причем второе приближенное равенство следует из предположения о быстром установлении равновесия. При этом предположении соотношение продуктов тогда равно
- .
![{\displaystyle {\frac {[\mathbf {D} ]}{[\mathbf {C} ]}}\approx {\frac {d[\mathbf {D} ]}{dt}}{\Big /}{ \frac {d[\mathbf {C} ]}{dt}}={\frac {k_{2}[\mathbf {B} ]}{k_{1}[\mathbf {A} ]}}\approx { \frac {k_{2}K[\mathbf {A} ]}{k_{1}[\mathbf {A} ]}}={\frac {k_{2}K}{k_{1}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/802efbeb75055ff826c7417118672230e54b99a2)
Другими словами, поскольку уравновешивание происходит быстрее по сравнению с образованием продукта, на протяжении всей реакции. Как результат, также остается примерно постоянным на протяжении всей реакции. В свою очередь, интегрирование по времени означает, что также принимает примерно постоянное значение в ходе реакции, а именно .
![{\displaystyle [\mathbf {B}]/[\mathbf {A} ]\приблизительно K}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e2f3778641d32c658e4c1ab2961f8735a102b329)
![{\displaystyle {\frac {d[\mathbf {D} ]}{dt}}{\Big /}{\frac {d[\mathbf {C} ]}{dt}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4f5d950d8350d10e6de7d872eea6986bbdd08b21)
![{\displaystyle [\mathbf {D} ]/[\mathbf {C} ]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/89143f8cee66e6e3cc5c89dea98fc4b2d65c8bc0)
Таким образом, с точки зрения энергий основного состояния и переходного состояния соотношение продуктов можно записать как:
- .
![{\displaystyle {\frac {[\mathbf {D} ]}{[\mathbf {C} ]}}\approx {\frac {k_{2}K}{k_{1}}} = {\frac {e ^{-\Delta G_{2}^{\ddagger }/RT}e^{-\Delta G^{\circ }/RT}}{e^{-\Delta G_{1}^{\ddagger }/ RT}}}=\exp {\big (}-(\Delta G_{2}^{\ddagger }-\Delta G_{1}^{\ddagger }+\Delta G^{\circ })/RT{ \большой )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b18fc40f30584687fab6672e87c8c07bc0160936)
Важно отметить, что рассмотрение приведенной выше энергетической диаграммы позволяет нам определить

с разницей энергий переходных состояний, что дает нам упрощенное уравнение, отражающее суть принципа Кертина-Хаммета:
![{\displaystyle {\frac {[\mathbf {D} ]}{[\mathbf {C} ]}}\approx e^{-\Delta \Delta G^{\ddagger }/RT}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c0d4bbae9e76a67c396d9d0cc15dc87bab82b7c6)
Таким образом, хотя соотношение продуктов зависит от константы равновесия между A и B и разницы в энергии между барьерами от A до C и от B до D , оба эти фактора автоматически учитываются разностью энергий переходных состояний. что приводит к продуктам ΔΔ G ‡ .
реакций под контролем Кертина Хэммета – Классы
Три основных класса реакций можно объяснить принципом Кертина-Хаммета: либо более или менее стабильный конформер может реагировать быстрее, либо они оба могут реагировать с одинаковой скоростью.
Более стабильный конформер реагирует быстрее I Случай :
Одна категория реакций под контролем Кертина-Хаммета включает превращения, при которых более стабильный конформер реагирует быстрее. Это происходит, когда состояние перехода от основного промежуточного продукта к соответствующему продукту имеет более низкую энергию, чем состояние перехода от второстепенного промежуточного продукта к другому возможному продукту. В этом случае основной продукт получается из основного конформера, и распределение продукта не отражает равновесное распределение конформеров.
Пример: окисление пиперидина [ править ]
Пример сценария Кертина-Хаммета, в котором более стабильный конформационный изомер реагирует быстрее, наблюдается при окислении пиперидинов . В случае N-метилпиперидина инверсия азота между диастереомерными конформерами происходит намного быстрее, чем скорость окисления амина. [6] Конформация, помещающая метильную группу в экваториальное положение, на 3,16 ккал/моль более стабильна, чем аксиальная конформация. [7] Соотношение продуктов 95:5 указывает на то, что более стабильный конформер дает основной продукт. [8]

Менее стабильный конформер реагирует быстрее II Случай :
Вторая категория реакций под контролем Кертина-Хэммета включает реакции, в которых менее стабильный конформер реагирует быстрее. В этом случае, несмотря на энергетическое предпочтение менее реакционноспособных частиц, основной продукт получается из частиц с более высокой энергией. Важным следствием является то, что продукт реакции может быть получен из конформера, который имеет достаточно низкую концентрацию, чтобы его нельзя было наблюдать в основном состоянии. [3]
тропановое Пример алкилирование :
Алкилирование . тропанов конформации йодидом метила является классическим примером сценария Кертина-Хэммета, в котором основной продукт может возникнуть из менее стабильной [3] Здесь менее стабильный конформер реагирует через более стабильное переходное состояние с образованием основного продукта. [9] Следовательно, конформационное распределение основного состояния не отражает распределение продуктов.

скоростью III: оба конформера реагируют с одинаковой . Случай
Гипотетически возможно, что два разных конформера, находящихся в равновесии, могут реагировать через переходные состояния, равные по энергии. В этом случае селективность продукта будет зависеть только от распределения конформеров в основном состоянии. В этом случае оба конформера будут реагировать с одинаковой скоростью.
Пример: реакция S N 2 циклогексилиодида [ править ]
Эрнест Л. Элиэль предположил, что гипотетическая реакция циклогексилиодида с радиоактивно меченным йодидом приведет к полностью симметричному переходному состоянию. [10] Поскольку как экваториальные, так и аксиально-замещенные конформеры будут реагировать через одно и то же переходное состояние, ΔΔG ‡ будет равен нулю. Согласно принципу Кертина-Хэммета, распределение продуктов должно быть на 50% аксиально-замещенным и на 50% экваториально-замещенным. Однако уравновешивание продуктов не позволяет наблюдать это явление. [3]

: радикальное Пример метилирование
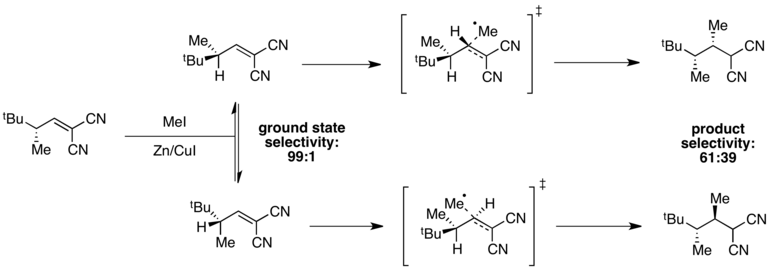
Когда энергии основного состояния различны, но энергии переходного состояния одинаковы, селективность в переходном состоянии будет ухудшаться, и может наблюдаться плохая общая селективность. Например, высокая селективность по одному конформеру основного состояния наблюдается в следующей реакции радикального метилирования . [11]

Конформер, в котором деформация A(1,3) минимальна, находится на минимуме энергии, что обеспечивает селективность 99:1 в основном состоянии. Однако энергии переходных состояний зависят как от наличия деформации A(1,3), так и от стерических затруднений, связанных с поступающим метильным радикалом. В этом случае эти два фактора находятся в оппозиции, и разница в энергиях переходных состояний мала по сравнению с разницей в энергиях основного состояния. В результате в реакции наблюдается плохая общая селективность.
к стереоселективным и региоселективным Применение реакциям
Принцип Кертина-Хаммета используется для объяснения коэффициентов селективности некоторых стереоселективных реакций.
к динамическому кинетическому Применение разрешению
Принцип Кертина-Хэммета может объяснить наблюдаемую динамику превращений с использованием динамического кинетического разрешения , таких как асимметричное гидрирование Нойори. [12] и энантиоселективное литиирование. [13]
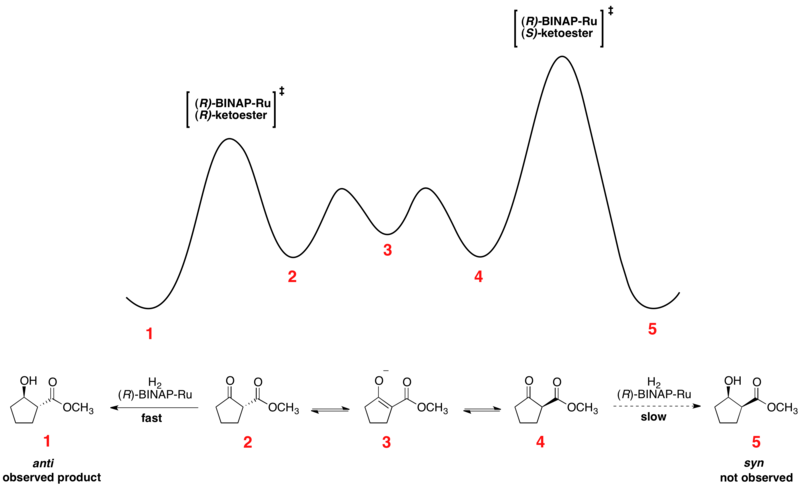
гидрирование Асимметричное Ноёри
Быстрое равновесие между энантиомерными конформерами и необратимое гидрирование ставят реакцию под контроль Кертина-Хаммета. Использование хирального катализатора с более высокой и более низкой энергией приводит к переходному состоянию для гидрирования двух энантиомеров. Преобразование происходит через переходное состояние с более низкой энергией с образованием продукта в виде одного энантиомера. [14] В соответствии с принципом Кертина-Хаммета соотношение продуктов зависит от абсолютного энергетического барьера необратимой стадии реакции и не отражает равновесное распределение конформеров субстрата. Относительный профиль свободной энергии одного примера асимметричного гидрирования Нойори показан ниже:
Энантиоселективное литиирование [ править ]
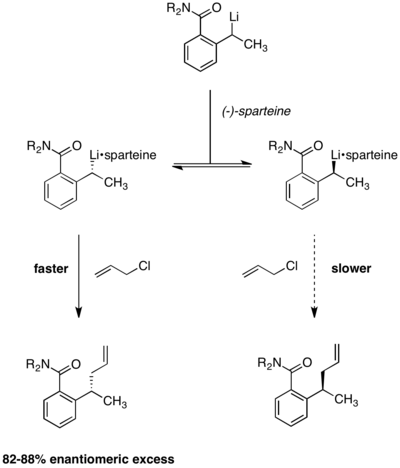
Динамическое кинетическое разрешение в условиях Куртина – Хэммета также применялось к реакциям энантиоселективного литиирования. В приведенной ниже реакции было замечено, что энантиоселективность продукта не зависит от хиральности исходного материала. Использование (-)-спартеина имеет важное значение для энантиоселективности, поскольку рацемический продукт. в его отсутствие образуется [13] Равновесие между двумя алкиллитиевыми комплексами было продемонстрировано наблюдением, что энантиоселективность оставалась постоянной в ходе реакции. Если бы два реагентных комплекса не преобразовывались быстро друг в друга, энантиоселективность со временем снижалась бы по мере истощения более быстро реагирующего конформера.
к региоселективному ацилированию Применение
Принцип Кертина-Хаммета был использован для объяснения региоселективности при ацилировании 1,2-диолов. Обычно менее затрудненный участок асимметричного 1,2-диола подвергается более быстрой этерификации из-за уменьшения стерических затруднений между диолом и ацилирующим реагентом. Развитие селективной этерификации наиболее замещенной гидроксильной группы является полезным преобразованием в синтетической органической химии, особенно в синтезе углеводов и других полигидроксилированных соединений. [15] Для эффективного достижения этого преобразования использовались ацетали станнилена. [16]

Асимметричный диол сначала обрабатывают оловянным реагентом для получения дибутилстаннилена ацеталя. Затем это соединение обрабатывают одним эквивалентом ацилхлорида для получения моноэфира станнила. Доступны два изомера станнилового эфира, которые могут подвергаться быстрому взаимному превращению через тетраэдрическое промежуточное соединение. Первоначально преобладает менее стабильный изомер, так как он быстрее образуется из станнилацеталя. Однако достижение равновесия между двумя изомерами приводит к избытку более стабильного первичного алкоксистаннана в растворе. Затем реакция необратимо гасится, при этом менее затрудненный первичный алкокси станнан реагирует быстрее. Это приводит к селективному производству более замещенного моноэфира. Это сценарий Кертина-Хэммета, в котором более стабильный изомер также реагирует быстрее.

к асимметричному эпоксидированию Применение
Эпоксидирование асимметричных алкенов также изучалось как пример кинетики Кертина – Хэммета. В вычислительном исследовании диастереоселективного эпоксидирования хиральных аллильных спиртов пероксикомплексами титана рассчитанная разница в энергиях переходных состояний между двумя конформерами составила 1,43 ккал/моль. [17] Экспериментально наблюдаемое соотношение продуктов составляло 91:9 в пользу продукта, полученного из переходного состояния с более низкой энергией. Это соотношение продуктов согласуется с вычисленной разницей в энергиях переходного состояния. Это пример, в котором конформер, предпочитаемый в основном состоянии, который испытывает пониженную деформацию A (1,3), реагирует через переходное состояние с более низкой энергией с образованием основного продукта.

Синтетические приложения [ править ]
Синтез AT2433-A1 [ править ]
Принцип Кертина-Хэммета был использован для объяснения селективности в различных синтетических путях. Один из примеров наблюдается на пути к противоопухолевому антибиотику AT2433-A1, в котором циклизация типа Манниха протекает с превосходной региоселективностью. Исследования показывают, что стадия циклизации необратима в растворителе, используемом для проведения реакции, что позволяет предположить, что кинетика Кертина-Хаммета может объяснить селективность продукта. [18]

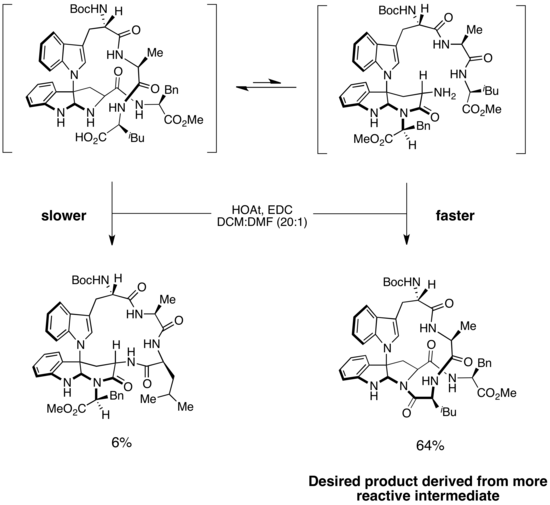
Синтез капакахинов B и F [ править ]
Сценарий Кертина-Хэммета был использован для объяснения селективности в синтезе капакахинов B и F, двух циклических пептидов, выделенных из морских губок. В структуре каждого из двух соединений присутствует скрученный 16-членный макроцикл. [19] Ключевым этапом синтеза является селективное образование амидной связи для образования правильного макроцикла. В энантиоселективном синтезе капакахинов B и F Фила Бэрана было предложено образование макроцикла происходить за счет двух изомеров субстрата. [20] Более легко доступный изомер с более низкой энергией привел к образованию нежелательного продукта, тогда как менее стабильный изомер образовал желаемый продукт. Однако поскольку стадия образования амидной связи была необратимой, а барьер изомеризации был низким, основной продукт был получен из более быстро реагирующего промежуточного продукта. Это пример сценария Кертина-Хэммета, в котором менее стабильное промежуточное соединение значительно более реакционноспособно, чем более стабильное промежуточное соединение, которое преобладает в растворе. Поскольку изомеризация субстрата происходит быстро, на протяжении всей реакции избыток субстрата более стабильной формы может превращаться в менее стабильную форму, которая затем подвергается быстрому и необратимому образованию амидной связи с образованием желаемого макроцикла. Эта стратегия обеспечила получение желаемого продукта с селективностью >10:1. (Я думаю, что в схеме ошибка. См. страницы обсуждения.)
Синтез (+ гризеофульвина - )
В первом энантиоселективном синтезе (+)-гризеофульвина , мощного противогрибкового средства, [21] наблюдалась ситуация Кертина-Хэммета. Ключевой стадией синтеза является катализируемое родием образование илида оксония, который затем подвергается [2,3]-сигматропной перегруппировке на пути к желаемому продукту. [22] Однако в субстрате присутствуют две орто-алкоксигруппы, каждая из которых предположительно может участвовать в генерации оксонийилида.
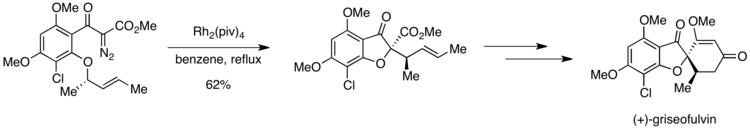
Однако добиться высокой селективности по целевому продукту удалось благодаря различиям в активационных барьерах на стадии, следующей за образованием илида. Если орто-метоксигруппа подвергается образованию илида оксония, 1,4-метиловый сдвиг может привести к образованию нежелательного продукта. Илид оксония, образованный из другой орто-алкоксигруппы, подвергается [2,3]-сигматропной перегруппировке с получением желаемого соединения. Пиррунг и его коллеги сообщили о полной селективности в отношении желаемого продукта по сравнению с продуктом, полученным в результате 1,4-метилового сдвига. Этот результат предполагает, что образование илида оксония обратимо, но последующий этап необратим. Разрешенная симметрией [2,3]-сигматропная перегруппировка должна идти по пути с более низкой энергией активации, чем 1,4-метиловый сдвиг, что объясняет исключительное образование желаемого продукта.

Синтез (+)-аллоциатина B 2 [ править ]
Потенциальный сценарий Куртина-Хэммета также был обнаружен во время энантиоселективного тотального синтеза (+)-аллоциатина B2 группой Троста. [23] Ключевой стадией синтеза стала диастереоселективная циклоизомеризация, катализируемая Ru. Реакция может привести к образованию двух возможных изомеров двойной связи. Реакция обеспечила хорошую селективность в отношении желаемого изомера, а результаты соответствовали сценарию Кертина-Хэммета. Первоначальное окислительное циклорутенирование и отщепление бета-гидрида дают винилрутенийгидрид. Вставка гидрида позволяет легко провести изомеризацию алкенов. Маловероятно, что результат реакции отражает стабильность промежуточных продуктов, поскольку большая группа CpRu испытывает неблагоприятные стерические взаимодействия с соседней изопропильной группой. Вместо этого применяется ситуация Кертина-Хэммета, в которой изомер, находящийся в равновесии, не приводит к образованию основного продукта. Восстановительному удалению предпочтительнее более реакционноспособное и менее стабильное промежуточное соединение, поскольку в переходном состоянии максимально снижается напряжение. В результате образуется желаемый изомер двойной связи.

См. также [ править ]
Ссылки [ править ]
- ^ Кэри, Фрэнсис А.; Сундберг, Ричард Дж.; (1984). Расширенная органическая химия. Часть A. Структура и механизмы (2-е изд.). Нью-Йорк, штат Нью-Йорк: Пленум Пресс. ISBN 0-306-41198-9
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (1994) « Принцип Кертина – Хэммета ». два : 10.1351/goldbook.C01480
- ↑ Перейти обратно: Перейти обратно: а б с д и Джеффри И. Симан (1983). «Влияние конформационных изменений на реакционную способность в органической химии. Оценки, применение и расширение кинетики Кутина – Хэммета/Винштейна – Холнесса». Химические обзоры . 83 (2): 83–134. дои : 10.1021/cr00054a001 .
- ^ Джеффри И. Симан (1986). «Принцип Кертина-Хэммета и уравнение Уинстейна-Холнесса». Журнал химического образования . 63 (1): 42–48. Бибкод : 1986ЖЧЭд..63...42С . дои : 10.1021/ed063p42 .
- ^ Всорек, Джозеф (18 декабря 2009 г.). «Принцип Кертина-Хэммета и уравнение Уинстейна-Холнесса» (PDF) . Семинары группы Эванс . Архивировано из оригинала (PDF) 18 сентября 2017 г. Проверено 19 ноября 2017 г.
- ^ Пи Джей Кроули; МДжТ Робинсон; М.Г. Уорд (1977). «Конформационные эффекты в соединениях с 6-членными кольцами-XII». Тетраэдр . 33 (9): 915–925. дои : 10.1016/0040-4020(77)80202-0 .
- ^ Луис Карбалейра; Игнасио Перес-Жюсте (1998). «Влияние уровня расчета и влияние метилирования на аксиальное/экваториальное равновесие в пиперидинах». Журнал вычислительной химии . 19 (8): 961–976. doi : 10.1002/(SICI)1096-987X(199806)19:8<961::AID-JCC14>3.0.CO;2-A .
- ^ Ю. Шво; ЭД Кауфман (1972). «Конфигурационный и конформационный анализ оксидов циклических аминов». Тетраэдр . 28 (3): 573–580. дои : 10.1016/0040-4020(72)84021-3 .
- ^ Родни Д. Отценбергер; Кеннет Б. Липковиц; Брэдфорд П. Манди (1974). «Кватернизации в ряду 8-азабицикло[4.3.0]нон-3-ена». Журнал органической химии . 39 (3): 319–321. дои : 10.1021/jo00917a008 .
- ^ Элиэль, Эрнест Л. (1962). Стереохимия углеродных соединений . Нью-Йорк: МакГроу-Хилл. стр. 149–156 , 234–239.
- ^ Гизе, Б.; Коппинг, Б.; Гобель, Т.; Дикхо, Дж.; Тома, Г.; Кулике, К.; Трач, Ф. (2004). Органические реакции .
- ^ М. Китамура; М. Токунага; Р. Нойори (1993). «Количественное выражение динамического кинетического разрешения хирально-лабильных энантиомеров: стереоселективное гидрирование 2-замещенных эфиров 3-оксокарбоновых кислот, катализируемое комплексами BINAP-рутений (II)». Журнал Американского химического общества . 115 (1): 144–152. дои : 10.1021/ja00054a020 .
- ↑ Перейти обратно: Перейти обратно: а б Питер Бик; Амит Басу; Дональд Дж. Галлахер; Ён Сан Пак; С. Таюманаван (1996). «Региоселективные, диастереоселективные и энантиоселективные последовательности литиирования-замещения: пути реакции и синтетические применения». Отчеты о химических исследованиях . 29 (11): 552–560. дои : 10.1021/ar950142b .
- ^ Ноёри, Рёдзи ; Икеда, Т.; Окума, Т.; Видхальм, М.; Китамура, М.; Такая, Х.; Акутагава, С.; Сайо, Н.; Сайто, Т.; Такетоми, Т.; Кумобаяшис, Х. (1989). «Стереоселективное гидрирование посредством динамического кинетического разрешения». Журнал Американского химического общества . 111 (25): 9134–9135. дои : 10.1021/ja00207a038 .
- ^ Уистлер, РЛ; Вольфром, М.Л. (1963). Методы химии углеводов . Академическая пресса .
- ^ Роленс, С. (1996). «Оловоорганическое моноацилирование диолов с обратной хемоселективностью». Журнал органической химии . 61 (16): 5257–5263. дои : 10.1021/jo960453f .
- ^ Кюи, М.; Адам, В.; Шен, Дж. Х.; Луо, XM; Тан, Х, Дж.; Чен, KX; Джи, Р.Ю.; Цзян, Х.Л. (2002). «Плотностно-функциональное исследование механизма диастереоселективного эпоксидирования хиральных аллиловых спиртов пероксидными комплексами титана». Журнал органической химии . 67 (5): 1427–1435. дои : 10.1021/jo016015c . ПМИД 11871869 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Чисхолм, доктор медицинских наук; Ван Вранкен, DL (2000). «Региоконтролируемый синтез противоопухолевого антибиотика AT2433-A1». Журнал органической химии . 65 (22): 7541–7553. дои : 10.1021/jo000911r . ПМИД 11076613 .
- ^ Накао, Ёичи; Юнг, Брайан К.С.; Ёсида, Уэсли Ю.; Шойер, Пол Дж.; Келли-Борхес, Мишель (1995). «Капакахин B, циклический гексапептид с альфа-карболиновой кольцевой системой из морской губки Cribrochalina olemda». Журнал Американского химического общества . 117 (31): 8271–8272. дои : 10.1021/ja00136a026 . ISSN 0002-7863 .
- ^ Ньюхаус, Т.; Льюис, Калифорния; Баран, П.С. (2009). «Энантиоспецифический общий синтез капакахинов B и F». Журнал Американского химического общества . 131 (18): 6360–6361. дои : 10.1021/ja901573x . ПМИД 19374357 .
- ^ Дэвис, Р.Р. (1980). Противогрибковая химиотерапия . Уайли и сыновья .
- ^ Пиррунг, MC; Браун, Уильям, Л.; Реге, С.; Лотон, П. (1991). «Тотальный синтез (+)-гризеофульвина». Журнал Американского химического общества . 113 (22): 8561–8562. дои : 10.1021/ja00022a075 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Трост, Б.М.; Донг, Л.; Шредер, генеральный директор (2005). «Тотальный синтез (+)-аллоциатина B 2 ». Журнал Американского химического общества . 127 (9): 2844–2845. дои : 10.1021/ja0435586 . ПМИД 15740107 .