Теория переходного состояния
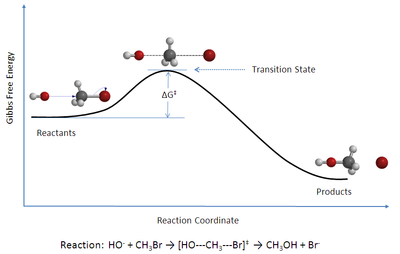
В химии ТСТ теория переходного состояния ( ) объясняет скорость элементарных химических реакций . Теория предполагает особый тип химического равновесия (квазиравновесия) между реагентами и активированными переходного состояния . комплексами [1]
TST используется в первую очередь для качественного понимания того, как происходят химические реакции. TST оказался менее успешным в своей первоначальной цели расчета абсолютных констант скорости реакции, поскольку расчет абсолютных скоростей реакции требует точного знания поверхностей потенциальной энергии . [2] но ему удалось рассчитать стандартную энтальпию активации (Δ H ‡ , также пишется Δ ‡ ЧАС ɵ ), стандартная энтропия активации (Δ S ‡ или Δ ‡ С ɵ ) и стандартную Гиббса энергию активации (Δ G ‡ или Δ ‡ Г ɵ ) для конкретной реакции, если ее константа скорости определена экспериментально. ( ‡ обозначение относится к процентной стоимости в переходном состоянии ; Δ Н ‡ представляет собой разницу между энтальпией переходного состояния и энтальпией реагентов.)
Эта теория была разработана одновременно в 1935 году Генри Айрингом , работавшим тогда в Принстонском университете , и Мередит Гвинн Эванс и Майклом Поланьи из Манчестерского университета . [3] [4] [5] TST также называют «теорией активированных комплексов», «теорией абсолютной скорости» и «теорией абсолютных скоростей реакций». [6]
До разработки TST закон скорости Аррениуса широко использовался для определения энергии барьера реакции. Уравнение Аррениуса выведено на основе эмпирических наблюдений и игнорирует любые механистические соображения, например, участвует ли один или несколько реакционноспособных промежуточных продуктов в превращении реагента в продукт. [7] связанных с этим законом: предэкспоненциальный множитель ( A ) и энергию активации ( Ea Следовательно, необходима дальнейшая разработка, чтобы понять два параметра , ). TST, который привел к уравнению Айринга , успешно решает эти две проблемы; однако между публикацией закона скорости Аррениуса в 1889 году и уравнением Эйринга, полученным на основе TST, в 1935 году прошло 46 лет. В течение этого периода многие ученые и исследователи внесли значительный вклад в развитие теории.
Теория [ править ]
Основные идеи теории переходного состояния заключаются в следующем:
- Скорость реакции можно изучить, исследуя активированные комплексы вблизи седловой точки поверхности потенциальной энергии . Детали формирования этих комплексов не важны. Сама седловая точка называется переходным состоянием.
- Активированные комплексы находятся в особом равновесии (квазиравновесии) с молекулами реагирующих веществ.
- Активированные комплексы могут превращаться в продукты, и с помощью кинетической теории можно рассчитать скорость этого превращения.
Развитие [ править ]
При разработке TST были использованы три подхода, которые кратко изложены ниже.
Термодинамическая обработка [ править ]
В 1884 году Якобус ван 'т-Гофф предложил уравнение Ван'т-Гоффа, описывающее температурную зависимость константы равновесия обратимой реакции:


где Δ U — изменение внутренней энергии, K — константа равновесия реакции, R — универсальная газовая постоянная , а T — термодинамическая температура . На основании экспериментальных работ в 1889 году Сванте Аррениус предложил аналогичное выражение для константы скорости реакции, задаваемое следующим образом:

Интегрирование этого выражения приводит к уравнению Аррениуса

где k — константа скорости. А называли частотным фактором (теперь называемым предэкспоненциальным коэффициентом), а считали Еа энергией активации. К началу 20 века многие приняли уравнение Аррениуса, но физическая интерпретация A и E a оставалась неясной. Это побудило многих исследователей химической кинетики предложить различные теории того, как происходят химические реакции, в попытке связать A и E a с молекулярной динамикой, непосредственно ответственной за химические реакции. [ нужна ссылка ]
В 1910 году французский химик Рене Марселин представил концепцию стандартной энергии активации Гиббса. Его отношение можно записать как

Примерно в то же время, когда Марселин работал над своей формулой, голландские химики Филип Абрахам Конштамм, Франс Эппо Корнелис Шеффер и Видольд Франс Брандсма ввели стандартную энтропию активации и стандартную энтальпию активации. Они предложили следующее уравнение константы скорости

Однако природа константы до сих пор оставалась неясной.
кинетической теорией Лечение
В начале 1900 года Макс Траутц и Уильям Льюис изучали скорость реакции с помощью теории столкновений , основанной на кинетической теории газов . Теория столкновений рассматривает реагирующие молекулы как твердые сферы, сталкивающиеся друг с другом; эта теория пренебрегает изменениями энтропии, поскольку предполагает, что столкновения между молекулами полностью упругие.
Льюис применил свою обработку к следующей реакции и получил хорошее согласие с экспериментальным результатом.
2HI → Н 2 + Я 2
Однако позже, когда тот же подход был применен к другим реакциям, возникли большие расхождения между теоретическими и экспериментальными результатами.
Статистико-механическая обработка [ править ]
Статистическая механика сыграла значительную роль в развитии ТСТ. Однако применение статистической механики к TST развивалось очень медленно, учитывая тот факт, что в середине 19 века Джеймс Клерк Максвелл , Людвиг Больцман и Леопольд Пфаундлер опубликовали несколько статей, в которых обсуждалось равновесие и скорости реакций с точки зрения молекулярных движений и статистического распределения. молекулярных скоростей.
Лишь в 1912 году французский химик А. Берту использовал закон распределения Максвелла-Больцмана для получения выражения для константы скорости.

где a и b — константы, связанные с энергетическими условиями.
Два года спустя Рене Марслен внес существенный вклад, рассматривая ход химической реакции как движение точки в фазовом пространстве . Затем он применил статистико-механические процедуры Гиббса и получил выражение, подобное тому, которое он получил ранее из термодинамических соображений.
В 1915 году еще один важный вклад сделал британский физик Джеймс Райс. На основании своего статистического анализа он пришел к выводу, что константа скорости пропорциональна «критическому приращению». Его идеи были далее развиты Ричардом Чейсом Толменом . В 1919 году австрийский физик Карл Фердинанд Герцфельд применил статистическую механику к константе равновесия и кинетическую теорию к константе скорости обратной реакции k -1 для обратимой диссоциации двухатомной молекулы. [8]
![{\displaystyle {\ce {AB <=>[k_1][k_{-1}] {A}+ {B}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8df558c371c7f125f5833608e30f847abe2601de)
Он получил следующее уравнение для константы скорости прямой реакции [9]

где – энергия диссоциации при абсолютном нуле, k B – постоянная Больцмана , h – постоянная Планка , T – термодинамическая температура, – частота колебаний связи.Это выражение очень важно, поскольку впервые коэффициент k B T / h , который является критическим компонентом TST, появляется в уравнении скорости.


В 1920 году американский химик Ричард Чейс Толман развил идею Райс о критическом приращении. Он пришел к выводу, что критическое приращение (теперь называемое энергией активации) реакции равно средней энергии всех молекул, вступающих в реакцию, минус средняя энергия всех реагирующих молекул.
энергетические Потенциальные поверхности
Концепция поверхности потенциальной энергии сыграла очень важную роль в разработке TST. Основы этой концепции были заложены Рене Марселеном в 1913 году. Он предположил, что ход химической реакции можно описать как точку на поверхности потенциальной энергии с координатами в атомных импульсах и расстояниях.
В 1931 году Генри Айринг и Майкл Поланьи построили поверхность потенциальной энергии для реакции, представленной ниже. Эта поверхность представляет собой трехмерную диаграмму, основанную на квантово-механических принципах, а также экспериментальных данных о частотах колебаний и энергиях диссоциации.
Ч + Ч 2 → Ч 2 + Ч
Через год после строительства Эйринга и Поланьи Ганс Пельцер и Юджин Вигнер внесли важный вклад, проследив за ходом реакции на поверхности потенциальной энергии. Важность этой работы заключалась в том, что впервые обсуждалась концепция кол или седловой точки на поверхности потенциальной энергии. Они пришли к выводу, что скорость реакции определяется движением системы через этот столбик.
Обычно предполагалось, что лимитирующая скорость или самая низкая седловая точка расположена на той же энергетической поверхности, что и исходное основное состояние. Однако недавно было обнаружено, что это может быть неверно для процессов, происходящих в полупроводниках и изоляторах, где исходное возбужденное состояние может проходить через седловую точку ниже, чем точка на поверхности исходного основного состояния. [10]
реакции Теория Крамерса скорости
Моделируя реакции как движение Ланжевена вдоль одномерной координаты реакции, Хендрик Крамерс смог вывести связь между формой поверхности потенциальной энергии вдоль координаты реакции и скоростями перехода системы. Формулировка основана на аппроксимации потенциального энергетического ландшафта как серии гармонических колодцев. В двухгосударственной системе будет три колодца; колодец для состояния А, перевернутый колодец, представляющий потенциальный энергетический барьер, и колодец для состояния Б.
В сверхзатухающем (или «диффузионном») режиме скорость перехода из состояния A в B связана с резонансной частотой ям соотношением

где - частота ямы для состояния A, – частота барьерной ямы, вязкое демпфирование, – энергия вершины барьера, - энергия забоя ямы для состояния A, а — температура системы, умноженная на постоянную Больцмана. [11]






Для общего демпфирования (чрезмерного или недостаточного демпфирования) существует аналогичная формула. [12]
Айринга уравнения Обоснование
Одной из наиболее важных особенностей, введенных Эйрингом , Поланьи и Эвансом, было представление о том, что активированные комплексы находятся в квазиравновесии с реагентами. Тогда скорость прямо пропорциональна концентрации этих комплексов, умноженной на частоту ( k B T / h ), с которой они превращаются в продукты. Ниже приводится нестрогая аргументация правдоподобия функциональной формы уравнения Эйринга. Однако ключевой статистико-механический коэффициент k B T / h не будет оправдан, а представленная ниже аргументация не представляет собой истинного «вывода» уравнения Эйринга. [13]
квазиравновесии Предположение о
Квазиравновесие отличается от классического химического равновесия, но может быть описано с использованием аналогичного термодинамического подхода. [6] [14] Рассмотрим реакцию ниже
![{\displaystyle {\ce {{A}+{B}<=>{[AB]^{\ddagger }}->{P}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0b873373ba74f1671f87574af29e3a0a9ba9c63d)
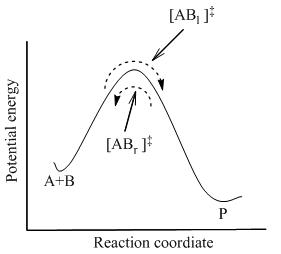
где достигается полное равновесие между всеми видами в системе, включая активированные комплексы, [AB] ‡ . Используя статистическую механику, концентрация [AB] ‡ можно рассчитать через концентрацию А и В.
TST предполагает, что даже когда реагенты и продукты не находятся в равновесии друг с другом, активированные комплексы находятся в квазиравновесии с реагентами. Как показано на рисунке 2, в любой момент времени существует несколько активированных комплексов, причем некоторые из них в ближайшем прошлом были молекулами-реагентами, которые обозначены [AB l ] ‡ (поскольку они движутся слева направо). Остальные из них были молекулами-продуктами в ближайшем прошлом ([AB r ] ‡ ).
В ТСТ предполагается, что поток активированных комплексов в двух направлениях не зависит друг от друга. То есть, если бы все молекулы продукта были внезапно удалены из реакционной системы, поток [AB r ] ‡ останавливается, но поток слева направо все еще существует. Следовательно, если быть технически корректным, реагенты находятся в равновесии только с [AB l ] ‡ , активированные комплексы, которые в недавнем прошлом были реагентами.
Аргумент правдоподобия
Активированные комплексы не подчиняются больцмановскому распределению энергий, но «константу равновесия» все же можно вывести из распределения, которому они следуют. Константа равновесия K ‡ для квазиравновесия можно записать как
- .
![{\displaystyle K^{\ddagger }={\frac {\ce {[AB]^{\ddagger }}}{\ce {[A][B]}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1465c535ba1f38d0eb964550468ee6c1f19aeb98)
Итак, химическая активность переходного состояния AB ‡ является
- .
![{\displaystyle [{\ce {AB}}]^{\ddagger }=K^{\ddagger }[{\ce {A}}][{\ce {B}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a67224b747342a2c608a96509499f845d991e801)
Следовательно, уравнение скорости производства продукции имеет вид
- ,
![{\displaystyle {\frac {d[{\ce {P}}]}{dt}}=k^{\ddagger }[{\ce {AB}}]^{\ddagger }=k^{\ddagger } K^{\ddagger }[{\ce {A}}][{\ce {B}}]=k[{\ce {A}}][{\ce {B}}]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/618074b91e2c1707bac08640100ceed7c7da2214)
где константа скорости k определяется выражением
- .

Вот, к ‡ прямо пропорциональна частоте колебательной моды, ответственной за превращение активированного комплекса в продукт; частота этой колебательной моды равна . Любая вибрация не обязательно приводит к образованию продукта, поэтому константа пропорциональности , называемый коэффициентом передачи, вводится для учета этого эффекта. Итак, к ‡ можно переписать как

- .

Для константы равновесия K ‡ , статистическая механика приводит к выражению, зависящему от температуры, задаваемому как
- ( ).


Комбинируя новые выражения для k ‡ и К ‡ , можно записать новое выражение для константы скорости, которое имеет вид
- .

Поскольку по определению ∆ G ‡ = Δ Н ‡ – Т Δ С ‡ выражение константы скорости можно расширить, чтобы получить альтернативную форму уравнения Эйринга:
- .

Для правильной размерности уравнение должно иметь дополнительный коэффициент ( c ⊖ ) 1– м для реакций, которые не являются мономолекулярными:
- ,

где с ⊖ стандартная концентрация 1 моль⋅л −1 m . – молекулярность [15]
из TST и связь с Аррениуса Выводы теорией
Выражение константы скорости из теории переходного состояния можно использовать для расчета Δ G ‡ , Δ H ‡ , ΔS ‡ и даже ∆ V ‡ (объем активации) с использованием экспериментальных данных о скорости. Эти так называемые параметры активации дают представление о природе переходного состояния , включая содержание энергии и степень порядка, по сравнению с исходными материалами и стали стандартным инструментом для выяснения механизмов реакций в физической органической химии . Свободная энергия активации, Δ G ‡ , определяется в теории переходного состояния как энергия такая, что держит. Параметры Δ H ‡ и Δ S ‡ затем можно сделать вывод, определив Δ G ‡ = Δ Н ‡ – Т Δ С ‡ при разных температурах.

Поскольку функциональная форма уравнений Эйринга и Аррениуса аналогична, возникает соблазн связать параметры активации с энергией активации и предэкспоненциальными коэффициентами лечения Аррениуса. Однако уравнение Аррениуса было получено на основе экспериментальных данных и моделирует макроскопическую скорость с использованием только двух параметров, независимо от количества переходных состояний в механизме. Напротив, параметры активации можно найти для каждого переходного состояния многоступенчатого механизма, по крайней мере в принципе. Таким образом, хотя энтальпия активации Δ H ‡ , часто приравнивают к энергии активации Аррениуса E a , они не эквивалентны. Для стадии реакции в конденсированной фазе (например, в фазе раствора) или мономолекулярной газофазной реакции E a = Δ H ‡ + РТ . Для других газофазных реакций E a = Δ H ‡ + (1 − Δ n ‡ ) RT , где ∆ n ‡ – изменение числа молекул при образовании переходного состояния. [16] (Таким образом, для бимолекулярного газофазного процесса E a = Δ H ‡ + 2 РТ. )
Энтропия активации, Δ S ‡ , показывает степень, в которой переходное состояние (включая любые молекулы растворителя, участвующие в реакции или возмущенные ею) является более разупорядоченным по сравнению с исходными материалами. Он предлагает конкретную интерпретацию предэкспоненциального множителя A в уравнении Аррениуса; для мономолекулярного одностадийного процесса грубая эквивалентность A = ( k B T / h ) exp(1 + Δ S ‡ / R ) (или A = ( k B T / h ) exp(2 + Δ S ‡ / R ) для бимолекулярных газофазных реакций). Для мономолекулярного процесса отрицательное значение указывает на более упорядоченное и жесткое переходное состояние, чем основное состояние, а положительное значение отражает переходное состояние с более слабыми связями и/или большей конформационной свободой. Важно отметить, что из соображений размерности реакции, которые являются бимолекулярными или выше, имеют Δ S ‡ значения, которые зависят от выбранного стандартного состояния (в частности, стандартной концентрации). В последних публикациях 1 моль л −1 или выбран 1 моляр. Поскольку этот выбор является человеческим конструктом, основанным на наших определениях единиц молярного количества и объема, величина и знак Δ S ‡ ибо одна реакция бессмысленна сама по себе; действительны только сравнения значения со значением эталонной реакции «известного» (или предполагаемого) механизма, выполненного в том же стандартном состоянии. [17]
Объем активации находится путем взятия частной производной от Δ G ‡ по давлению (при постоянной температуре): . Он дает информацию о размере и, следовательно, степени связи в переходном состоянии. Ассоциативный механизм, скорее всего, будет иметь отрицательный объем активации, тогда как диссоциативный механизм, скорее всего, будет иметь положительное значение.

Учитывая связь между константой равновесия и константами скорости вперед и назад, , уравнение Эйринга означает, что

- .

Еще одним следствием TST является принцип Кертина-Хаммета : соотношение продуктов кинетически контролируемой реакции от R к двум продуктам A и B будет отражать разницу в энергиях соответствующих переходных состояний, приводящих к продукту, при условии, что существует единственный переход. сказать каждому:
- ( ).
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=e^{-\Delta \Delta G^{\ddagger }/RT}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac8276860d8cfb32f02aba8aba55acef114beb8b)

(В выражении для ΔΔ G ‡ выше есть доп. термин, если A и B образованы из двух разных видов SA и SB , находящихся в равновесии.)

Для термодинамически контролируемой реакции каждая разница RT ln 10 ≈ (1,987 × 10 −3 ккал/моль K)(298 K)(2,303) ≈ 1,36 ккал/моль в свободной энергии продуктов A и B приводит к 10-кратному увеличению селективности при комнатной температуре (298 K), принцип, известный как «правило 1,36». ":
- ( ).
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=10^{-\Delta G^{\circ }/(1,36\ \mathrm {ккал/моль}) }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/91b01377882f2cca09a5e47e866e2f82b0a9a222)

Аналогично, каждая разница в свободной энергии активации на 1,36 ккал/моль приводит к десятикратному увеличению селективности кинетически контролируемого процесса при комнатной температуре: [18]
- ( ).
![{\displaystyle {\frac {[\mathrm {A} ]}{[\mathrm {B} ]}}=10^{-\Delta \Delta G^{\ddagger }/(1,36\ \mathrm {ккал/моль) } )}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b17890458cfb78285761eed11143f5343072c979)

Используя уравнение Эйринга, существует прямая связь между Δ G ‡ , константы скорости первого порядка и период полураспада реакции при данной температуре. При 298 К протекает реакция с Δ G ‡ = 23 ккал/моль имеет константу скорости k ≈ 8,4 × 10 −5 с −1 полуторапериод t 1/2 ≈ 2,3 часа, цифры часто округляют до k ~ 10 −4 с −1 и t 1/2 ~ 2 ч. Таким образом, свободная энергия активации такой величины соответствует типичной реакции, завершающейся в течение ночи при комнатной температуре. Для сравнения, переворот стула из циклогексана имеет Δ G ‡ около 11 ккал/моль при k ~ 10 5 с −1 , что делает его динамическим процессом, который происходит быстро (быстрее, чем время ЯМР) при комнатной температуре. На другом конце шкалы цис/транс- изомеризация 2-бутена имеет Δ G ‡ около 60 ккал/моль, что соответствует k ~ 10 −31 с −1 при 298 К. Это ничтожная скорость: период полураспада на 12 порядков превышает возраст Вселенной . [19]
Ограничения [ править ]
В целом, TST предоставил исследователям концептуальную основу для понимания того, как происходят химические реакции. Несмотря на то, что теория широко применима, она имеет ограничения. Например, применительно к каждому элементарному этапу многостадийной реакции теория предполагает, что каждое промежуточное соединение достаточно долгоживущее, чтобы достичь распределения энергий Больцмана, прежде чем перейти к следующему этапу. Когда промежуточные соединения очень недолговечны, TST терпит неудачу. [20] В таких случаях импульс траектории реакции от реагентов к промежуточному продукту может продолжаться, что влияет на селективность продукта. Примером такой реакции является замыкание цикла циклопентановых бирадикалов, образующихся при газофазном термическом разложении 2,3-диазабицикло[2.2.1]гепт-2-ена. [21] [22]
Теория переходного состояния также основана на предположении, что атомные ядра ведут себя в соответствии с классической механикой . [23] Предполагается, что реакция не происходит, если атомы или молекулы не сталкиваются с достаточной энергией для образования переходной структуры. Однако, согласно квантовой механике, для любого барьера с конечным количеством энергии существует вероятность того, что частицы все равно смогут туннелировать через барьер. Применительно к химическим реакциям это означает, что существует вероятность того, что молекулы вступят в реакцию, даже если они не столкнутся с достаточной энергией, чтобы преодолеть энергетический барьер. [24] Хотя этот эффект незначителен для реакций с большими энергиями активации, он становится важным явлением для реакций с относительно низкими энергетическими барьерами, поскольку вероятность туннелирования увеличивается с уменьшением высоты барьера.
Теория переходного состояния не работает для некоторых реакций при высокой температуре. Теория предполагает, что реакционная система пройдет через седловую точку с самой низкой энергией на поверхности потенциальной энергии. Хотя это описание соответствует реакциям, протекающим при относительно низких температурах, при высоких температурах молекулы занимают колебательные моды с более высокой энергией; их движение становится более сложным, и столкновения могут привести к переходным состояниям далеко от седловой точки с самой низкой энергией. Это отклонение от теории переходного состояния наблюдается даже в простой реакции обмена между двухатомным водородом и водородным радикалом. [25]
Учитывая эти ограничения, было предложено несколько альтернатив теории переходного состояния. Далее следует краткое обсуждение этих теорий.
переходного Обобщенная состояния теория
Любая форма TST, такая как микроканонический вариационный TST, канонический вариационный TST и улучшенный канонический вариационный TST, в которых переходное состояние не обязательно расположено в седловой точке, называется обобщенной теорией переходного состояния.
TST Микроканонический вариационный
Фундаментальный недостаток теории переходного состояния заключается в том, что она считает любое пересечение переходного состояния реакцией реагентов на продукты или наоборот. В действительности молекула может пересечь эту «разделительную поверхность» и развернуться, или пересечь ее несколько раз и реагировать по-настоящему только один раз. Таким образом, считается, что нескорректированный TST обеспечивает верхнюю границу коэффициентов скорости. Чтобы исправить это, вариационная теория переходного состояния изменяет расположение разделительной поверхности, которая определяет успешную реакцию, чтобы минимизировать скорость для каждой фиксированной энергии. [26] Выражения скорости, полученные при такой микроканонической трактовке, можно проинтегрировать по энергии с учетом статистического распределения по энергетическим состояниям, чтобы получить канонические или тепловые скорости.
Канонический вариационный TST [ править ]
Развитие теории переходного состояния, в которой положение разделительной поверхности изменяется так, чтобы минимизировать константу скорости при заданной температуре.
TST канонический Улучшенный вариационный
Модификация канонической вариационной теории переходного состояния, в которой для энергий ниже пороговой энергии положение разделительной поверхности считается положением микроканонической пороговой энергии. Это приводит к тому, что вклады в константы скорости равны нулю, если они ниже пороговой энергии. Затем выбирается компромиссная разделительная поверхность, чтобы минимизировать вклад в константу скорости реагентов, имеющих более высокие энергии.
Неадиабатический TST [ править ]
Расширение TST на реакции, в которых одновременно участвуют два спиновых состояния, называется теорией неадиабатического переходного состояния (NA-TST) .
Полуклассический TST [ править ]
Используя теорию колебательных возмущений, такие эффекты, как туннелирование и вариационные эффекты, можно объяснить в рамках формализма SCTST .
Приложения [ править ]
Ферментативные реакции [ править ]
Ферменты катализируют химические реакции с поразительной скоростью по сравнению с некаталитической химией при тех же условиях реакции. Каждое каталитическое событие требует минимум трех или часто более стадий, все из которых происходят в течение нескольких миллисекунд, которые характеризуют типичные ферментативные реакции. Согласно теории переходного состояния, наименьшая часть каталитического цикла тратится на самую важную стадию — переходное состояние. Первоначальные предложения теории абсолютной скорости химических реакций определяли переходное состояние как отдельный вид в координате реакции, которая определяет абсолютную скорость реакции. Вскоре после этого Лайнус Полинг предположил, что мощное каталитическое действие ферментов можно объяснить специфическим прочным связыванием с видами переходного состояния. [27] Поскольку скорость реакции пропорциональна доле реагента в комплексе переходного состояния, было предложено использовать фермент для увеличения концентрации реакционноспособных веществ.
Это предложение было формализовано Вулфенденом и его коллегами из Университета Северной Каролины в Чапел-Хилл , которые предположили, что увеличение скорости, вызываемое ферментами, пропорционально сродству фермента к структуре переходного состояния по отношению к комплексу Михаэлиса. [28] Поскольку ферменты обычно увеличивают скорость некаталитической реакции в 10 раз. 6 -10 26 и комплексы Михаэлиса [ нужны разъяснения ] часто имеют константы диссоциации в пределах 10 −3 -10 −6 M предполагается, что комплексы переходного состояния связаны с константами диссоциации в диапазоне 10 −14 -10 −23 М. По мере того как субстрат превращается из комплекса Михаэлиса в продукт, химический процесс происходит за счет индуцированных ферментами изменений в распределении электронов в субстрате. Ферменты изменяют электронную структуру путем протонирования, отрыва протона, переноса электрона, геометрического искажения, гидрофобного разделения и взаимодействия с кислотами и основаниями Льюиса. Таким образом, аналоги, которые напоминают структуры переходного состояния, должны представлять собой самые мощные из известных нековалентных ингибиторов.
Все химические превращения проходят через нестабильную структуру, называемую переходным состоянием, которая находится между химическими структурами субстратов и продуктов. Предполагается, что переходные состояния для химических реакций имеют время жизни около 10 −13 секунд, порядка времени колебания одинарной связи. Не существует физического или спектроскопического метода для непосредственного наблюдения структуры переходного состояния ферментативных реакций, однако структура переходного состояния имеет центральное значение для понимания ферментативного катализа, поскольку ферменты работают за счет снижения энергии активации химического превращения.
В настоящее время принято, что функция ферментов заключается в стабилизации переходных состояний, лежащих между реагентами и продуктами, и что, следовательно, можно ожидать, что они будут прочно связывать любой ингибитор, который очень похож на такое переходное состояние. Субстраты и продукты часто участвуют в нескольких реакциях, катализируемых ферментами, тогда как переходное состояние обычно характерно для одного конкретного фермента, поэтому такой ингибитор имеет тенденцию быть специфичным для этого конкретного фермента. Идентификация многочисленных ингибиторов переходного состояния подтверждает гипотезу стабилизации переходного состояния для ферментативного катализа.
В настоящее время известно большое количество ферментов, взаимодействующих с аналогами переходного состояния, большинство из которых были разработаны с целью ингибирования целевого фермента. Примеры включают протеазу ВИЧ-1, рацемазы, β-лактамазы, металлопротеиназы, циклооксигеназы и многие другие.
на поверхностях и реакции поверхностях на Адсорбция
Десорбцию, а также реакции на поверхностях легко описать с помощью теории переходного состояния. Анализ адсорбции на поверхности из жидкой фазы может представлять собой проблему из-за отсутствия возможности оценить концентрацию растворенного вещества вблизи поверхности. Когда полные сведения недоступны, было предложено нормализовать концентрации реагирующих веществ по концентрации активных участков поверхности - приближение, называемое приближением равной плотности поверхностных реагентов (SREA). [29]
См. также [ править ]
Примечания [ править ]
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Теория переходного состояния ». два : 10.1351/goldbook.T06470
- ^ Трулар, Д.Г.; Гарретт, Британская Колумбия; Клиппенштейн, С.Дж. (1996). «Текущее состояние теории переходного состояния». Дж. Физ. Хим . 100 (31): 12771–12800. дои : 10.1021/jp953748q .
- ^ М. Г. Эванс, М. Поланьи (1935), «Некоторые применения метода переходного состояния для расчета скоростей реакций, особенно в растворе» , Труды Общества Фарадея , том. 31, стр. 875–894, doi : 10.1039/tf9353100875 , ISSN 0014-7672 , получено 7 мая 2024 г.
- ^ Лейдлер, К.; Кинг, К. (1983). «Развитие теории переходного состояния». Дж. Физ. Хим . 87 (15): 2657. doi : 10.1021/j100238a002 .
- ^ Лейдлер, К.; Кинг, К. (1998). «Жизнь теории переходного состояния». Химический разведчик . 4 (3): 39.
- ↑ Перейти обратно: Перейти обратно: а б Лейдлер, К.Дж. (1969). Теории скорости химических реакций . МакГроу-Хилл.
- ^ Анслин, Э.В.; Догерти, Д.А. (2006). «Теория переходного состояния и смежные темы». Современная физико-органическая химия . Университетские научные книги. стр. 365–373. ISBN 1891389319 .
- ^ Герцфельд, К.Э. (1919). «К теории скоростей реакций в газах» . Анналы физики . 364 (15): 635–667. Бибкод : 1919АнП...364..635Х . дои : 10.1002/andp.19193641504 . S2CID 122909496 .
- ^ Кейт Дж. Лейдлер , Химическая кинетика (3-е изд., Harper & Row 1987), стр.88 ISBN 0-06-043862-2
- ^ Луо, Г.; Куеч, Т.Ф.; Морган, Д. (2018). «Окислительно-восстановительное состояние переходного состояния при динамических процессах в полупроводниках и изоляторах». Материалы НПГ Азия . 10 (4): 45–51. arXiv : 1712.01686 . Бибкод : 2018npgAM..10...45L . дои : 10.1038/s41427-018-0010-0 . S2CID 67780897 .
- ^ Линдси, Стюарт (2010). Введение в нанонауку . Издательство Оксфордского университета. стр. 109–111.
- ^ «23.2: Теория Крамерса» . Химия LibreTexts . 17 января 2021 г. Проверено 11 июня 2023 г.
- ^ Вводное описание статистической механики и элементарный вывод уравнения Айринга см.: Лоури и Ричардсон, Механизм и теория в органической химии , 3-е изд. (Харпер и Роу, 1987), стр. 248–253.
- ^ Стейнфельд, Джеффри Л.; Франциско, Джозеф С.; Хейс, Уильям Л. (1999). Химическая кинетика и динамика (2-е изд.). Прентис-Холл. стр. 289–293. ISBN 0-13-737123-3 .
- ^ Лейдлер, Кейт Дж. (1981). «Символика и терминология химической кинетики» (PDF) . Чистая и прикладная химия . 53 . ИЮПАК: 753–771 . Проверено 9 августа 2019 г.
См. стр.765, примечание м.
- ^ Стейнфельд, Джеффри Л.; Франциско, Джозеф С.; Хейс, Уильям Л. (1999). Химическая кинетика и динамика (2-е изд.). Прентис-Холл. п. 302. ИСБН 0-13-737123-3 .
- ^ Карпентер, Барри К. (1984). Определение механизмов органических реакций . Нью-Йорк: Уайли. ISBN 0471893692 . ОСЛК 9894996 .
- ^ Лоури, Томас Х. (1987). Механизм и теория в органической химии . Ричардсон, Кэтлин Шуллер. (3-е изд.). Нью-Йорк: Харпер и Роу. ISBN 0060440848 . OCLC 14214254 .
- ^ Элиэль, Эрнест Л. (Эрнест Людвиг) (1994). Стереохимия органических соединений . Вилен, Сэмюэл Х., Мандер, Льюис Н. Нью-Йорк: Wiley. ISBN 0471016705 . ОСЛК 27642721 .
- ^ Карпентер, Барри К. (31 декабря 1998 г.). «Динамическое поведение органических реактивных промежуточных продуктов». Angewandte Chemie, международное издание . 37 (24): 3340–3350. doi : 10.1002/(SICI)1521-3773(19981231)37:24<3340::AID-ANIE3340>3.0.CO;2-1 . eISSN 1521-3773 . ISSN 1433-7851 . ПМИД 29711290 .
- ^ Догерти, Деннис А.; Анслин, Эрик В. (2006). Современная физико-органическая химия . Саусалито, Калифорния, США: Университетские научные книги. п. 374. ИСБН 9781891389313 .
- ^ Рейес, Майра Б.; Карпентер, Барри К. (1 октября 2000 г.). «Механизм термической деазетизации 2,3-диазабицикло[2.2.1]гепт-2-ена и динамика его реакции в сверхкритических флюидах». Журнал Американского химического общества . 122 (41): 10163–10176. дои : 10.1021/ja0016809 . eISSN 1520-5126 . ISSN 0002-7863 .
- ^ Айринг, Х. (1935). «Активированный комплекс в химических реакциях». Дж. Хим. Физ . 3 (2): 107–115. Бибкод : 1935ЖЧФ...3..107Е . дои : 10.1063/1.1749604 .
- ^ Мазель, Р. (1996). Принципы адсорбции и реакций на твердых поверхностях . Нью-Йорк: Уайли.
- ^ Пинеда, младший; Шварц, С.Д. (2006). «Динамика белков и катализ: проблемы теории переходного состояния и тонкости динамического контроля» . Фил. Пер. Р. Сок. Б. 361 (1472): 1433–1438. дои : 10.1098/rstb.2006.1877 . ПМЦ 1647311 . ПМИД 16873129 .
- ^ Трулар, Д.; Гаррет, Б. (1984). «Вариационная теория переходного состояния» . Анну. Преподобный физ. Хим . 35 : 159–189. Бибкод : 1984ARPC...35..159T . дои : 10.1146/annurev.pc.35.100184.001111 .
- ^ Полинг, Л. (1948). «Химические достижения и надежда на будущее». Американский учёный . 36 (1): 50–58. ПМИД 18920436 .
- ^ Радзичка, А.; Вулфенден, Р. (1995). «Опытный фермент». Наука . 267 (5194): 90–93. Бибкод : 1995Sci...267...90R . дои : 10.1126/science.7809611 . ПМИД 7809611 .
- ^ Дойл, Питер Дж.; Савара, Адитья; Райман, Стивен С. (2020). «Извлечение значимых стандартных энтальпий и энтропий активации поверхностных реакций из кинетических скоростей» . Кинетика, механизмы и катализ реакций . 129 (2): 551–581. дои : 10.1007/s11144-020-01747-2 . ОСТИ 1606821 . S2CID 211836011 .
Ссылки [ править ]
- Анслин, Эрик В.; Доутери, Деннис А., Теория переходного состояния и смежные темы. В научных книгах Университета современной физической органической химии : 2006; стр. 365–373
- Клеланд, В.В., Изотопные эффекты: определение структуры переходного состояния фермента. Методы энзимологии 1995, 249, 341–373.
- Лейдлер, К.; Кинг, К., Развитие теории переходного состояния. Журнал физической химии 1983, 87, (15), 2657.
- Лейдлер К. Жизнь теории переходного состояния. Химический разведчик 1998, 4, (3), 39
- Радзичка, А.; Волденден Р. Переходное состояние и мультисубстратные аналоговые ингибиторы. Методы энзимологии 1995, 249, 284–312.
- Шрамм, В.Л., Ферментативные переходные состояния и аналоговый дизайн переходного состояния. Ежегодный обзор биохимии 1998, 67, 693–720.
- Шрамм В.Л., Теория ферментативного переходного состояния и разработка аналога переходного состояния. Журнал биологической химии 2007, 282, (39), 28297-28300.