Электрофильное ароматическое замещение
Электрофильное ароматическое замещение ( SE , Ar ) — органическая реакция в которой атом, присоединенный к ароматической системе (обычно водороду), заменяется электрофилом . Некоторыми из наиболее важных электрофильных ароматических замещений являются ароматическое нитрование , ароматическое галогенирование , ароматическое сульфирование алкилирования , реакция Фриделя-Крафтса и реакция ацилирования Фриделя-Крафтса. [1]
Показательные реакции [ править ]
Наиболее широко практикуемым примером этой реакции является этилирование бензола.

В 1999 году было произведено около 24 700 000 тонн. [2] товарный пластиковый полистирол (После дегидрирования и полимеризации производится .) В этом процессе кислоты используются в качестве катализатора для образования зарождающегося карбокатиона. Проводятся многие другие электрофильные реакции бензола, хотя и в гораздо меньших масштабах; они являются ценными маршрутами к ключевым промежуточным звеньям. Нитрование бензола достигается за счет действия иона нитрония в качестве электрофила. Сульфирование дает дымящей серной кислотой бензолсульфоновую кислоту . Ароматическое галогенирование бромом хлором , . или йодом дает соответствующие арилгалогениды Эта реакция обычно катализируется соответствующим тригалогенидом железа или алюминия.

Реакцию Фриделя -Крафтса можно проводить либо как ацилирование , либо как алкилирование . Часто трихлорид алюминия используют практически любую сильную кислоту Льюиса , но можно применить . Для реакции ацилирования стехиометрическое требуется количество трихлорида алюминия.

Механизм реакции [ править ]
Общий механизм реакции, обозначенный механистическим символом Хьюза-Ингольда S E Ar , [3] начинается с атаки ароматического кольца на электрофил E + (2а). Этот этап приводит к образованию положительно заряженного и делокализованного циклогексадиенильного катиона , также известного как ион арения , интермедиат Уиланда или ареновый σ-комплекс (2b). множество примеров этого карбокатиона , но при нормальных условиях эксплуатации эти высококислотные виды отдают протон, присоединенный к sp. Охарактеризовано 3 углерод в растворитель (или любое другое слабое основание) для восстановления ароматичности. Конечным результатом является замена H на E в арильном кольце (3).
Иногда другие электрофуги (группы, которые могут уйти без своей электронной пары ). рядом с H появляются + уйдет, чтобы восстановить ароматичность; эти виды включают силильные группы (как SiR 3 + ), карбоксильная группа (как CO 2 + H + ), группа йода (как я + ) и третичные алкильные группы, такие как т -бутил (как R + ). Способность этих типов заместителей к удалению иногда используется синтетически, особенно в случае замены силила другой функциональной группой ( атака ipso ). Однако потеря таких групп, как йод или алкил, чаще всего является нежелательной побочной реакцией.

Влияние групп заместителей [ править ]

Как на региоселективность (различные модели замещения аренов ), так и на скорость электрофильного ароматического замещения влияют заместители, уже присоединенные к бензольному кольцу. С точки зрения региоселективности, некоторые группы способствуют замещению в орто- или пара -положениях, тогда как другие группы предпочитают замену в мета-положении. Эти группы называются либо орто-пара-направляющими , либо мета-направляющими соответственно. Кроме того, некоторые группы увеличивают скорость реакции ( активация ), а другие снижают скорость ( дезактивация ). Хотя закономерности региоселективности можно объяснить с помощью резонансных структур , влияние на кинетику можно объяснить как резонансными структурами , так и индуктивным эффектом .
Скорость реакции [ править ]
Заместители обычно можно разделить на два класса по электрофильному замещению: активирующие и дезактивирующие по направлению к ароматическому кольцу. Активирующие заместители или активирующие группы стабилизируют катионный промежуточный продукт, образующийся во время замещения, путем отдачи электронов в кольцевую систему либо за счет индуктивного эффекта , либо за счет резонансных эффектов . Примерами активированных ароматических колец являются толуол , анилин и фенол .
Дополнительная электронная плотность, вносимая в кольцо заместителем, не распределяется равномерно по всему кольцу, а концентрируется на атомах 2, 4 и 6, поэтому активирующие заместители также являются орто/пара-директорами (см. ниже).
С другой стороны, дезактивирующие заместители дестабилизируют промежуточный катион и, таким образом, снижают скорость реакции за счет индуктивного или резонансного эффекта. Они делают это, удаляя электронную плотность из ароматического кольца. Деактивация ароматической системы означает, что для завершения реакции обычно требуются более суровые условия. Примером этого является нитрование толуола при производстве тринитротолуола (ТНТ). Если первое нитрование, на активированном толуольном кольце, можно осуществить при комнатной температуре и разбавленной кислоте, то второе, на дезактивированном нитротолуольном кольце, уже требует длительного нагревания и более концентрированной кислоты, а третье - на очень сильно дезактивированном кольце. динитротолуола необходимо проводить в кипящей концентрированной серной кислоте . Группы, которые электроноакцепторуют за счет резонанса, уменьшают электронную плотность, особенно в позициях 2, 4 и 6, оставляя позиции 3 и 5 как обладающие сравнительно более высокой реакционной способностью, поэтому эти типы групп являются метадиректорами (см. Ниже). Галогены электроотрицательны, поэтому они дезактивируются путем индукции, но у них есть неподеленные пары, поэтому они являются донорами резонанса и, следовательно, орто/пара-директорами.
Орто/ парадиректора [ править ]
Группы с неподеленными парами электронов, такие как аминогруппа анилина , являются сильно активирующими (на некоторое время дезактивирующими также в случае галогенидов ) и орто/пара -направленными за счет резонанса. Такие активирующие группы отдают эти неподеленные электроны пи -системе, создавая отрицательный заряд в орто- и пара-положениях. Таким образом, эти позиции являются наиболее реактивными по отношению к бедным электронами электрофилам. Эта повышенная реакционная способность может быть компенсирована стерическими препятствиями между активирующей группой и электрофилом, но, с другой стороны, для реакции существует два орто-положения, но только одно пара-положение. Следовательно, конечный результат электрофильного ароматического замещения трудно предсказать, и обычно он устанавливается только путем проведения реакции и наблюдения за соотношением орто- и пара-замещения.

resonance structures for ortho attack of an electrophile on aniline

Помимо повышенной нуклеофильности исходного кольца, когда электрофил атакует орто- и пара-положения анилина, атом азота может отдавать электронную плотность пи- системе (образуя иминиум ), давая четыре резонансные структуры (в отличие от три в основной реакции). Это существенно повышает стабильность катионного промежуточного продукта.

Resonance structures for para attack

Когда электрофил атакует мета-положение, атом азота не может передать электронную плотность пи- системе, давая только три вкладчика в резонанс. Это рассуждение согласуется с низкими выходами метазамещенного продукта.

resonance structures for meta attack of an electrophile on aniline

Другие заместители, такие как алкильные и арильные заместители , также могут передавать электронную плотность пи -системе; однако, поскольку у них нет доступной неподеленной пары электронов, их способность делать это весьма ограничена. Таким образом, они лишь слабо активируют кольцо и не сильно ухудшают мета- позицию.
Направленное орто-металлирование — это особый тип ЭАС со специальными орто-директорами .
Мета- директора [ править ]
Негалогенные группы с атомами более электроотрицательными, чем углерод, например группа карбоновой кислоты (-CO 2 H), отнимают значительную электронную плотность из пи- системы. Эти группы являются сильно деактивирующими группами . Кроме того, поскольку замещенный углерод уже беден электронами, любая структура, имеющая резонансный вклад, в которой имеется положительный заряд на углероде, несущем электроноакцепторную группу (т.е. орто- или пара -атака), менее стабильна, чем другие. Следовательно, эти электроноакцепторные группы являются мета -направляющими, потому что это позиция, которая не имеет такой большой дестабилизации.
Реакция также протекает значительно медленнее (относительная скорость реакции 6×10 −8 по сравнению с бензолом), поскольку кольцо менее нуклеофильно.
Реакция на пиридин [ править ]
По сравнению с бензолом скорость электрофильного замещения пиридина значительно медленнее из-за более высокой электроотрицательности атома азота. Кроме того, азот в пиридине легко получает положительный заряд либо за счет протонирования (в результате нитрования или сульфирования ), либо за счет кислот Льюиса (таких как AlCl 3 ), используемых для катализа реакции. Это делает реакцию еще медленнее из-за наличия соседних формальных зарядов на углероде и азоте или двух формальных зарядов на локализованном атоме. Провести электрофильное замещение непосредственно в пиридине практически невозможно.
Чтобы осуществить реакцию, их можно провести двумя возможными реакциями, обе из которых являются косвенными.
Одним из возможных способов замещения пиридина является нуклеофильное ароматическое замещение. Даже в отсутствие катализаторов атом азота, будучи электроотрицательным, может сам удерживать отрицательный заряд. Другой способ - провести окисление перед электрофильным замещением. При этом образуется пиридина N -оксид , который за счет отрицательного атома кислорода делает реакцию быстрее, чем у пиридина и даже бензола. Затем оксид можно восстановить до замещенного пиридина.
Ипсо атака [ править ]
Присоединение входящей группы к положению ароматического соединения, уже несущему замещающую группу (кроме водорода). Входящая группа может вытеснить эту группу заместителей, но также может сама быть исключена или мигрировать в другое положение на последующем этапе. Термин « ipso -замещение» не используется, поскольку он является синонимом замены. [4] Классический пример — реакция салициловой кислоты со смесью азотной и серной кислот с образованием пикриновой кислоты . Нитрование 2-го положения приводит к потере CO 2 в качестве уходящей группы. Типичным примером является десульфонирование, при котором сульфонильная группа заменяется протоном. См. также перегруппировку Хаяши . В ароматических соединениях, замещенных кремнием, кремний реагирует путем замещения ipso .
Пятичленные гетероциклы [ править ]
По сравнению с бензолом фураны , тиофены и пирролы более подвержены электрофильному воздействию. Все эти соединения содержат атом с неподеленной парой электронов ( кислорода , серы или азота ) в качестве члена ароматического кольца, что существенно стабилизирует катионное промежуточное соединение. Примерами электрофильных замещений пиррола являются реакция Пикте-Шпенглера и реакция Бишлера-Наперальского .
ароматическое Асимметричное замещение электрофильное
Электрофильные ароматические замещения прохиральными углеродными электрофилами были адаптированы для асимметричного синтеза путем перехода на хиральные на основе кислот Льюиса, катализаторы особенно в реакциях типа Фриделя-Крафтса . Ранний пример касается присоединения хлораля к фенолам, катализируемого хлоридом алюминия, модифицированным (–)-ментолом . [5] Глиоксилатное с соединение было добавлено к N,N-диметиланилину хиральным бисоксазолиновым лигандом - каталитической системой трифлат меди (II), также в процессе гидроксиалкилирования Фриделя-Крафтса : [6]

Asymmetric Friedel–Crafts hydroxyalkylation

В другом алкилировании N-метилпиррол реагирует с кротональдегидом , катализируемым трифторуксусной кислотой, модифицированной хиральным имидазолидиноном : [7]
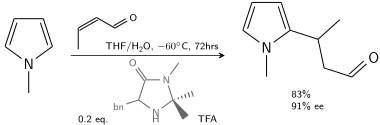
Friedel Crafts Asymmetric Addition To Pyrrole

Индол реагирует с енамидом, катализируемым хиральной БИНОЛа , производной фосфорной кислотой : [8]
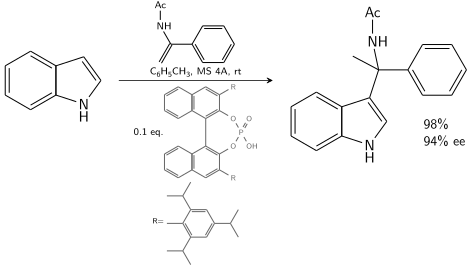
Friedel Crafts Alkylation Indole Asymmetric

В присутствии 10–20 % хирального катализатора 80–90 % ее достижима .
Другие реакции [ править ]
- Другие реакции, которые следуют схеме электрофильного ароматического замещения, представляют собой группу реакций ароматического формилирования, включая реакцию Вильсмайера-Хаака , реакцию Гаттермана-Коха и реакцию Реймера-Тимана .
- Другими электрофилами являются ароматические соли диазония в диазониевых сочетаниях , диоксид углерода в реакции Кольбе-Шмитта и активированные карбонильные группы в конденсации Пехмана , ион гидроксикарбения в хлорметилировании Блана через промежуточный (гидроксиметил)арен (бензиловый спирт), хлорильный катион (ClO 3 ) . + ) для электрофильного перхлорилирования .
- В многостадийной реакции Лемстедта-Танасеску одним из электрофилов является N-нитрозо-интермедиат.
- В реакции Черняка-Эйнхорна (названной в честь Джозефа Черняка и Альфреда Эйнхорна ) электрофил представляет собой N-метанольное производное амида . [9] [10]
См. также [ править ]
Ссылки [ править ]
- ^ Смит, Майкл Б.; Марч, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Винсент А. Уэлч, Кевин Дж. Фэллон, Хайнц-Питер Гельбке «Этилбензол», Энциклопедия промышленной химии Ульмана , Wiley-VCH, Вайнхайм, 2005. два : 10.1002/14356007.a10_035.pub2
- ^ Гоули, Роберт Э. (4 июня 1999 г.). «Предложение по (небольшой) модификации механистических дескрипторов Хьюза – Ингольда для реакций замещения» . Буквы тетраэдра . 40 (23): 4297–4300. дои : 10.1016/S0040-4039(99)00780-7 . ISSN 0040-4039 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « ipso -attack ». дои : 10.1351/goldbook.I03251
- ^ Асимметричное электрофильное замещение фенолов при гидроксиалкилировании Фриделя-Крафтса. Энантиоселективное ортогидроксиалкилирование, опосредованное хиральными хлоридами алкоксиалюминия Франка Биджи, Джованни Казираги, Джузеппе Каснати, Джованни Сартори, Джованна Гаспарри Фава и Мариса Феррари Беликки J. Org. хим. ; 1985 год ; 50(25) стр. 5018–5022; два : 10.1021/jo00225a003
- ^ Каталитические энантиоселективные реакции Фриделя-Крафтса ароматических соединений с глиоксилатом: простая процедура синтеза оптически активных эфиров ароматической миндальной кислоты Николас Гатергуд, Вэй Чжуан и Карл Анкер Йоргенсен Дж. Ам. хим. Соц. ; 2000 ; 122(50) стр. 12517–12522; (Статья) два : 10.1021/ja002593j
- ^ Новые стратегии в органическом катализе: первое энантиоселективное органокаталитическое алкилирование Фриделя-Крафтса Ник А. Парас и Дэвид В.К. Макмиллан Дж. Ам. хим. Соц. ; 2001 год ; 123(18) стр. 4370–4371; (Коммуникация) два : 10.1021/ja015717g
- ^ Энантиоселективная реакция Фриделя-Крафтса, катализируемая хиральной кислотой Бренстеда, индолов и альфа-арильных енамидов: построение четвертичных атомов углерода И-Ся Цзя, Цзюнь Чжун, Шоу-Фей Чжу, Кан-Мин Чжан и Ци-Лин Чжоу Ангью. хим. Межд. Эд. 2007 , 46, 5565 –5567 два : 10.1002/anie.200701067
- ^ Способ получения бензилфталимидов, Джозефа Черняка патент Германии , 1902 г. , DE-134,979.
- ^ О N-метилоловых соединениях амидов кислот [Первый трактат] Альфред Эйнхорн, Эдуард Бишкопф, Бруно Селински, Густав Шупп, Эдуард Шпренгертс, Карл Ладиш и Теодор Мауэрмайер Анналы Либигса 1905 , 343 , стр. 207–305 два : 10.1002/jlac.19053430207