S N 1 реакция

Реакция мономолекулярного нуклеофильного замещения ( SN ) 1 — реакция замещения в органической химии . Символ Хьюза-Ингольда в механизме выражает два свойства: «SN » означает « нуклеофильное замещение », а «1» говорит, что стадия, определяющая скорость, является мономолекулярной . [1] [2] Таким образом, уравнение скорости часто показано как имеющее зависимость первого порядка от субстрата и зависимость нулевого порядка от нуклеофила . Эта зависимость справедлива для ситуаций, когда количество нуклеофила намного больше, чем количество промежуточного продукта. Вместо этого уравнение скорости может быть более точно описано с использованием стационарной кинетики . В реакции участвует промежуточный карбокатион , и она обычно наблюдается в реакциях вторичных или третичных алкилгалогенидов в сильно основных условиях или, в сильно кислых условиях, с вторичными или третичными спиртами . С первичными и вторичными алкилгалогенидами протекает альтернативная реакция S N 2 . В неорганической химии реакцию S N 1 часто называют диссоциативным замещением . Этот путь диссоциации хорошо описывается цис-эффектом . Механизм реакции был впервые предложен Кристофером Ингольдом и др. в 1940 году. [3] Эта реакция мало зависит от силы нуклеофила, в отличие от механизма S N 2 . Этот тип механизма включает в себя два этапа. Первым этапом является ионизация алкилгалогенида в присутствии водного ацетона или этилового спирта. На этом этапе в качестве промежуточного продукта используется карбокатион.
На первом этапе механизма S N 1 образуется карбокатион, который является плоским, и, следовательно, атака нуклеофила (второй этап) может происходить с любой стороны с образованием рацемического продукта, но фактически полной рацемизации не происходит. Это связано с тем, что нуклеофильные частицы атакуют карбокатион еще до того, как уходящий ион галогенида отошел достаточно далеко от карбокатиона. Отрицательно заряженный галогенид-ион защищает карбокатион от атаки с передней стороны, а атака с обратной стороны, которая приводит к инверсии конфигурации, является предпочтительной. Таким образом, реальный продукт, несомненно, состоит из смеси энантиомеров, но энантиомеры с инвертированной конфигурацией будут преобладать, и полной рацемизации не происходит.
Механизм [ править ]
Примером реакции, протекающей по S N 1, механизму реакции является гидролиз трет -бутилбромида с образованием трет -бутанола :
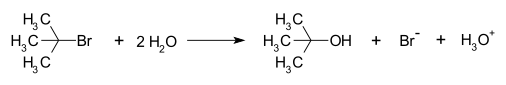
Эта реакция S N 1 протекает в три стадии:
- Образование трет -бутилкарбокатиона путем отделения уходящей группы ( бромид- аниона) от атома углерода: этот этап протекает медленно. [4]


Рекомбинация карбокатиона с нуклеофилом


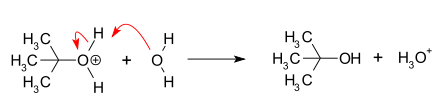
- Нуклеофильная атака: карбокатион реагирует с нуклеофилом. Если нуклеофил представляет собой нейтральную молекулу (т.е. растворитель ), для завершения реакции требуется третий этап. Когда растворителем является вода, промежуточным продуктом является ион оксония. Эта стадия реакции является быстрой.

- Депротонирование : Удаление протона у протонированного нуклеофила водой, действующей как основание, образующее спирт и ион гидроксония . Эта стадия реакции является быстрой.
Закон о ставках [ править ]
Хотя закон скорости реакции S N 1 часто рассматривается как имеющий первый порядок по алкилгалогениду и нулевой порядок по нуклеофилу, это упрощение справедливо только при определенных условиях. Хотя это тоже является приближением, закон скорости, полученный на основе приближения устойчивого состояния (SSA), позволяет лучше понять кинетическое поведение реакции S N 1. Рассмотрим следующую схему реакции по механизму, показанному выше:

-бутильный катион является относительно стабильным третичным карбокатионом , Несмотря на то, что трет он представляет собой высокоэнергетическую разновидность, которая присутствует только в очень низкой концентрации и не может наблюдаться непосредственно в нормальных условиях. Таким образом, SSA можно применить к этому виду:
- (1) Предположение об установившемся состоянии: d [ t Bu + ]/ dt = 0 = k 1 [ t BuBr] – k – 1 [ t Bu + ][Бр – ] – k 2 [ t Бу + ][Н 2 О]
- (2) Концентрация трет -бутильного катиона, исходя из предположения об устойчивом состоянии: [ t Bu + ] = k 1 [ t BuBr]/( k – 1 [Br – ] + к 2 [Н 2 О])
- (3) Общая скорость реакции при условии быстрой финальной стадии: d [ t BuOH]/ dt = k 2 [ t BuOH] + ][Н 2 О]
- (4) Стационарный закон скорости, подставив (2) в (3): d [ t BuOH]/ dt = k 1 k 2 [ t BuBr][H 2 O]/( k – 1 [Br – ] + к 2 [Н 2 О])
В обычных условиях синтеза входящий нуклеофил более нуклеофильен, чем уходящая группа, и присутствует в избытке. Кроме того, кинетические эксперименты часто проводятся в условиях начальной скорости (конверсия 5–10%) и без добавления бромида, поэтому [Br – ] ничтожно мало. По этим причинам k – 1 [Br – ] ≪ k 2 [H 2 O] часто имеет место. В этих условиях закон ставок SSA сводится к
скорость = d [ t BuOH]/ dt = k 1 k 2 [ t BuBr][H 2 O]/( k 2 [H 2 O]) = k 1 [ t BuBr],
простой закон скорости первого порядка, описанный во вводных учебниках. В этих условиях концентрация нуклеофила не влияет на скорость реакции, а смена нуклеофила (например, с H 2 O на MeOH) не влияет на скорость реакции, хотя продукт, конечно, другой. В этом режиме первая стадия (ионизация алкилбромида) протекает медленно, определяет скорость и необратима, а вторая стадия (нуклеофильное присоединение) протекает быстро и кинетически незаметно.
Однако при определенных условиях может наблюдаться кинетика реакции не первого порядка. В частности, когда присутствует большая концентрация бромида, а концентрация воды ограничена, обратный этап первой стадии становится важным кинетически. Как указывает закон скорости ССА, в этих условиях существует дробная (между нулевым и первым порядком) зависимость от [H 2 O], а от [Br] — отрицательная зависимость дробного порядка. – ]. S N Так, часто наблюдается замедление реакции 1 при добавлении к реакционной смеси экзогенного источника уходящей группы (в данном случае бромида). Это известно как эффект общего иона , и наблюдение этого эффекта свидетельствует о механизме S N 1 (хотя отсутствие эффекта общего иона не исключает этого). [5] [6]
Область применения [ править ]
Механизм S N 1 имеет тенденцию доминировать, когда центральный атом углерода окружен объемистыми группами, поскольку такие группы стерически препятствуют реакции S N 2. Кроме того, объемные заместители центрального углерода увеличивают скорость образования карбокатионов из-за снятия стерической деформации возникающей . Полученный карбокатион также стабилизируется как за счет индуктивной стабилизации, так и за счет гиперконъюгации присоединенных алкильных групп. Постулат Хаммонда-Леффлера предполагает, что это также увеличит скорость образования карбокатионов. Таким образом, механизм S N 1 доминирует в реакциях на третичных алкильных центрах.
Примером реакции, протекающей по типу S N 1, является синтез 2,5-дихлор-2,5-диметилгексана из соответствующего диола с концентрированной соляной кислотой : [7]

По мере увеличения количества альфа- и бета-замен по отношению к уходящим группам реакция переключается с S N 2 на S N 1.
Стереохимия [ править ]
Промежуточный карбокатион, образующийся на стадии определения скорости реакции (RDS), представляет собой sp 2 гибридизованный углерод с тригональной планарной молекулярной геометрией. Это допускает два разных способа нуклеофильной атаки: по одному с каждой стороны плоской молекулы. Если ни один из подходов не является предпочтительным, то эти два пути происходят одинаково, образуя рацемическую смесь энантиомеров, если реакция происходит в стереоцентре. [8] Это проиллюстрировано ниже на реакции SN 1 S-3-хлор-3-метилгексана с иодид-ионом, которая дает рацемическую смесь 3-йод-3-метилгексана:

Однако может наблюдаться избыток одного стереоизомера, поскольку уходящая группа может оставаться вблизи карбокатионного интермедиата в течение короткого времени и блокировать нуклеофильную атаку. Это контрастирует с механизмом S N 2 , который представляет собой стереоспецифический механизм, в котором стереохимия всегда инвертируется, когда нуклеофил входит с задней стороны уходящей группы.
Побочные реакции [ править ]
Двумя распространенными побочными реакциями являются реакции элиминирования и перегруппировка карбокатионов . Если реакция проводится в теплых или горячих условиях (которые способствуют увеличению энтропии), отщепление E1 вероятно, будет преобладать , что приведет к образованию алкена . При более низких температурах реакции S N 1 и E1 являются конкурирующими реакциями, и становится трудно отдать предпочтение одной из них. Даже если реакцию проводить в холодном состоянии, может образоваться некоторое количество алкена. Если попытаться провести реакцию S N 1 с использованием сильноосновного нуклеофила, такого как ион гидроксида или метоксида , алкен снова образуется, на этот раз посредством отщепления E2 . Это будет особенно верно, если реакция нагревается. Наконец, если промежуточный карбокатион может перегруппироваться в более стабильный карбокатион, это даст продукт, полученный из более стабильного карбокатиона, а не продукт простого замещения.
Эффекты растворителя
Поскольку реакция S N 1 включает образование нестабильного промежуточного карбокатиона на стадии определения скорости (RDS), все, что может облегчить этот процесс, ускорит реакцию. В качестве нормальных растворителей выбирают как полярные (для стабилизации ионных промежуточных продуктов в целом), так и протонные растворители (в частности, для сольватации уходящей группы). Типичные полярные протонные растворители включают воду и спирты, которые также действуют как нуклеофилы, и этот процесс известен как сольволиз.
Шкала Y коррелирует скорости реакции сольволиза любого растворителя ( k реакции стандартного растворителя (80% об/об этанол / вода ) ( k0 ) со скоростью ) через

где m - константа реагента (m = 1 для трет -бутилхлорида ) и Y - параметр растворителя. [9] Например, 100% этанол дает Y = -2,3, 50% этанол в воде Y = +1,65 и 15% концентрация Y = +3,2. [10]
См. также [ править ]
Ссылки [ править ]
- ^ Л.Г. Уэйд-младший, Органическая химия , 6-е изд., Пирсон/Прентис Холл, Аппер-Сэддл-Ривер, Нью-Джерси, США, 2005 г.
- ^ Марч, Дж. (1992). Продвинутая органическая химия (4-е изд.). Нью-Йорк: Уайли. ISBN 0-471-60180-2 .
- ^ Бейтман Л.К., Черч М.Г., Хьюз Э.Д., Ингольд К.К., Тахер Н.А. (1940). «188. Механизм замещения у насыщенного атома углерода. Часть XXIII. Кинетическая демонстрация мономолекулярного сольволиза алкилгалогенидов. (Раздел Е) общее обсуждение». Журнал Химического общества (обновленный) : 979. doi : 10.1039/JR9400000979 .
- ^ Петерс, К.С. (2007). «Природа динамических процессов, связанных с механизмом реакции SN1». хим. Откр. 107 (3): 859–873. дои : 10.1021/cr068021k . ПМИД 17319730 .
- ^ Анслин, Эрик В., 1960- (2006). Современная физическая органическая химия . Догерти, Деннис А., 1952-. Милл-Вэлли, Калифорния: Университетские научные книги. стр. 638–639. ISBN 1-891389-31-9 . OCLC 55600610 .
{{cite book}}: CS1 maint: несколько имен: список авторов ( ссылка ) CS1 maint: числовые имена: список авторов ( ссылка ) - ^ Лоури, Томас Х. (1987). Механизм и теория в органической химии . Ричардсон, Кэтлин Шуллер. (3-е изд.). Нью-Йорк: Харпер и Роу. стр. 330–331. ISBN 0-06-044084-8 . OCLC 14214254 .
- ^ Вагнер, Карл Э.; Маршалл, Памела А. (2010). «Синтез 2,5-дихлор-2,5-диметилгексана по реакции SN1». Дж. Хим. Образование. 87 (1): 81–83. Бибкод : 2010JChEd..87...81W . дои : 10.1021/ed8000057 .
- ^ Соррелл, Томас Н. «Органическая химия, 2-е издание», University Science Books, 2006 г.
- ^ Эрнест Грюнвальд и С. Винстейн (1948). «Корреляция скоростей сольволиза». Дж. Ам. хим. Соц. 70 (2): 846. doi : 10.1021/ja01182a117 .
- ^ Арнольд Х. Файнберг и С. Винстейн (1956). «Корреляция скоростей сольволиза. III.1 т-бутилхлорид в широком диапазоне смесей растворителей». Дж. Ам. хим. Соц . 78 (12): 2770. doi : 10.1021/ja01593a033 .
Внешние ссылки [ править ]
- Диаграммы : Фростбургский государственный университет
- Упражнение : Университет штата Мэн