Нуклеофильное ацильное замещение
Нуклеофильное ацильное замещение ( SN нуклеофилов Acyl описывает класс реакций замещения с участием ) и ацильных соединений. В реакциях этого типа нуклеофил, такой как спирт , амин или енолят , замещает уходящую группу ацильного производного, такого как галогенангидрид , ангидрид или сложный эфир . Полученный продукт представляет собой карбонилсодержащее соединение, в котором нуклеофил занимает место уходящей группы, присутствующей в исходном ацилпроизводном. Поскольку ацильные производные реагируют с широким спектром нуклеофилов и поскольку продукт может зависеть от конкретного типа ацильного производного и нуклеофила, реакции нуклеофильного ацильного замещения можно использовать для синтеза множества различных продуктов.
Механизм реакции
[ редактировать ]Карбонильные соединения реагируют с нуклеофилами по механизму присоединения: нуклеофил атакует карбонильный углерод, образуя тетраэдрический интермедиат . Эту реакцию можно ускорить в кислых условиях, которые делают карбонил более электрофильными , или в основных условиях, которые обеспечивают более анионный и, следовательно, более реакционноспособный нуклеофил. Тетраэдрический промежуточный продукт сам по себе может представлять собой спирт или алкоксид , в зависимости от pH реакции.
Тетраэдрическое промежуточное соединение ацильного соединения содержит заместитель, присоединенный к центральному углероду, который может действовать как уходящая группа . После образования тетраэдрического промежуточного продукта он разрушается, воссоздавая карбонильную связь C=O и выбрасывая уходящую группу в реакции элиминирования . В результате этого двухстадийного процесса присоединения/отщепления нуклеофил занимает место уходящей группы в карбонильном соединении через промежуточное состояние, не содержащее карбонил. Обе стадии обратимы , поэтому реакции нуклеофильного ацильного замещения являются равновесными процессами. [1] [ нужна полная цитата ] Поскольку равновесие будет благоприятствовать продукту, содержащему лучший нуклеофил, уходящая группа должна быть сравнительно плохим нуклеофилом, чтобы реакция была практической.
Кислые условия
[ редактировать ]В кислой среде карбонильная группа ацильного соединения 1 протонируется, что активирует его в направлении нуклеофильной атаки. На втором этапе протонированный карбонил 2 подвергается атаке нуклеофила (H-Z) с образованием тетраэдрического интермедиата 3 . Перенос протона от нуклеофила (Z) к уходящей группе (X) дает соединение 4 , которое затем разрушается, выбрасывая протонированную уходящую группу (H-X), давая протонированное карбонильное соединение 5 . Потеря протона дает продукт замещения 6 . Поскольку последний этап включает потерю протона, реакции нуклеофильного ацильного замещения считаются каталитическими в кислоте. Также обратите внимание, что в кислых условиях нуклеофил обычно существует в протонированной форме (т.е. H-Z вместо Z − ).

Основные условия
[ редактировать ]В основных условиях нуклеофил (Nuc) атакует карбонильную группу ацильного соединения 1 с образованием тетраэдрического алкоксидного промежуточного соединения 2 . Промежуточное соединение разрушается и вытесняет уходящую группу (X) с образованием продукта замещения 3 . Хотя реакции нуклеофильного ацильного замещения могут катализироваться основаниями, реакция не произойдет, если уходящая группа является более сильным основанием, чем нуклеофил (т. е. уходящая группа должна иметь более высокое значение p K a, чем нуклеофил). В отличие от процессов, катализируемых кислотами, как нуклеофил, так и уходящая группа существуют в виде анионов в основных условиях.

Этот механизм подтверждается экспериментами по мечению изотопов . Когда этилпропионат с этоксигруппой, меченной кислородом-18 , обрабатывается гидроксидом натрия (NaOH), метка кислорода-18 полностью отсутствует в пропионовой кислоте и обнаруживается исключительно в этаноле . [2]

Тенденции реактивности
[ редактировать ]Существует пять основных типов ацильных производных. Галогенангидриды являются наиболее реакционноспособными по отношению к нуклеофилам, за ними следуют ангидриды , сложные эфиры и амиды . Карбоксилат -ионы практически не реагируют на нуклеофильное замещение, поскольку не имеют уходящей группы. Реакционная способность этих пяти классов соединений охватывает широкий диапазон; относительные скорости реакций хлорангидридов и амидов различаются в 10 раз. 13 . [3]

Основным фактором, определяющим реакционную способность ацильных производных, является способность уходящей группы, которая связана с кислотностью. Слабые базы лучше покидают группы, чем сильные; вид с сильной сопряженной кислотой (например, соляная кислота ) будет лучшей уходящей группой, чем вид со слабой сопряженной кислотой (например, уксусная кислота ). Таким образом, хлорид -ион является лучшей уходящей группой, чем ацетат-ион . Реакционная способность ацильных соединений по отношению к нуклеофилам снижается с увеличением основности уходящей группы, как видно из таблицы. [4]
| Составное имя | Структура | Выход из группы | p K a сопряженной кислоты |
|---|---|---|---|
| Ацетилхлорид |  |  | −7 |
| Уксусный ангидрид |  |  | 4.76 |
| Этилацетат |  |  | 15.9 |
| Ацетамид |  |  | 38 |
| Ацетат- анион | | Н/д | Н/д |

Другим фактором, играющим роль в определении реакционной способности ацильных соединений, является резонанс . Амиды проявляют две основные резонансные формы. Оба вносят основной вклад в общую структуру, настолько, что амидная связь между карбонильным углеродом и амидным азотом имеет значительный характер двойной связи . Энергетический барьер вращения вокруг амидной связи составляет 75–85 кДж/моль (18–20 ккал/моль), что намного превышает значения, наблюдаемые для обычных одинарных связей. Например, связь C–C в этане имеет энергетический барьер всего 12 кДж/моль (3 ккал/моль). [3] Как только нуклеофил атакует и образуется тетраэдрический интермедиат, энергетически выгодный резонансный эффект теряется. Это помогает объяснить, почему амиды являются одними из наименее реакционноспособных ацильных производных. [4]
Эфиры обладают меньшей резонансной стабилизацией, чем амиды, поэтому образование тетраэдрического интермедиата и последующая потеря резонанса не так энергетически невыгодны. Ангидриды испытывают еще более слабую резонансную стабилизацию, поскольку резонанс разделен между двумя карбонильными группами, и они более реакционноспособны, чем сложные эфиры и амиды. В галогенангидридах резонанс очень мал, поэтому энергетические потери за образование тетраэдрического промежуточного соединения невелики. Это помогает объяснить, почему галогенангидриды являются наиболее реакционноспособными ацильными производными. [4]
Реакции ацильных производных
[ редактировать ]Многие реакции нуклеофильного ацильного замещения включают превращение одного ацильного производного в другое. В общем, чтобы быть практичными, превращения между ацильными производными должны происходить от относительно реакционноспособного соединения к менее реакционноспособному; хлорангидрид можно легко превратить в сложный эфир, но превращение сложного эфира непосредственно в хлорангидрид практически невозможно. При преобразовании между ацильными производными продукт всегда будет более стабильным, чем исходное соединение.
Возможны также реакции нуклеофильного ацильного замещения, не предполагающие взаимного превращения ацильных производных. Например, амиды и карбоновые кислоты реагируют с реактивами Гриньяра с образованием кетонов. Здесь представлен обзор реакций, в которых может участвовать каждый тип ацильных производных.
Галогенангидриды
[ редактировать ]Галогенангидриды являются наиболее реакционноспособными ацильными производными и легко превращаются в любые другие. Галогенангидриды реагируют с карбоновыми кислотами с образованием ангидридов. Если структура кислоты и хлорангидрида различна, продукт представляет собой смешанный ангидрид. Сначала карбоновая кислота атакует хлорангидрид ( 1 ) с образованием тетраэдрического интермедиата 2 . Тетраэдрическое промежуточное соединение разрушается, выбрасывая ион хлорида в качестве уходящей группы и образуя оксония разновидности 3 . Депротонирование дает смешанный ангидрид 4 и эквивалент HCl.

Спирты и амины реагируют с галогенангидридами с образованием сложных эфиров и амидов соответственно в реакции, формально известной как реакция Шоттена-Баумана . [5] Галогенангидриды гидролизуются в присутствии воды с образованием карбоновых кислот, но этот тип реакции редко бывает полезным, поскольку карбоновые кислоты обычно используются для синтеза галогенангидридов. Большинство реакций с галогенангидридами проводится в присутствии ненуклеофильного основания, такого как пиридин , для нейтрализации галоидоводородной кислоты, образующейся в качестве побочного продукта.
Галогенангидриды будут реагировать с углеродными нуклеофилами, такими как нуклеофилы Гриньяра и еноляты , хотя в результате могут образовываться смеси продуктов. Хотя углеродный нуклеофил сначала реагирует с галогенангидридом с образованием кетона, кетон также подвержен нуклеофильной атаке и может быть преобразован в третичный спирт. Например, когда бензоилхлорид ( 1 ) обрабатывают двумя эквивалентами реактива Гриньяра, такого как метилмагнийбромид (MeMgBr), 2-фенил-2-пропанол ( 3 ) получается с отличным выходом. Хотя ацетофенон ( 2 ) является промежуточным продуктом в этой реакции, его невозможно выделить, поскольку он быстро реагирует со вторым эквивалентом MeMgBr после образования. [6]


В отличие от большинства других углеродных нуклеофилов, диалкилкупраты лития, часто называемые реагентами Гилмана , могут присоединяться к галогенангидридам только один раз, образуя кетоны. Однако реакция между галоидангидридом и реагентом Гилмана не является реакцией нуклеофильного ацильного замещения и, как полагают, протекает по радикальному пути. [2] Синтез кетонов Вайнреба также можно использовать для преобразования галогенангидридов в кетоны. В этой реакции галоидангидрид сначала превращается в N-метокси-N-метиламид, известный как амид Вейнреба. Когда углеродный нуклеофил, такой как реактив Гриньяра или литийорганический реагент, присоединяется к амиду Вайнреба, металл хелатируется карбонильными и N-метоксикислородами, предотвращая дальнейшие нуклеофильные присоединения. [7]
При ацилировании Фриделя-Крафтса галогенангидриды действуют как электрофилы для электрофильного ароматического замещения . Кислота Льюиса , такая как хлорид цинка (ZnCl 2 ), хлорид железа (III) (FeCl 3 ) или хлорид алюминия (AlCl 3 ), координируется с галогеном галогенангидридной кислоты, активируя соединение в направлении нуклеофильной атаки активированным ароматическим соединением. кольцо. Для особенно богатых электронами ароматических колец реакция будет протекать без кислоты Льюиса. [8]
Тиоэфиры
[ редактировать ]Химический состав тиоэфиров и галогенангидридов аналогичен, реакционная способность напоминает хлорангидриды, но более мягкая.
Ангидриды
[ редактировать ]Химия галогенангидридов и ангидридов аналогична. Хотя ангидриды нельзя превратить в галогенангидриды, их можно превратить в остальные ацильные производные. Ангидриды также участвуют в реакциях типа Шоттена-Баумана с образованием сложных эфиров и амидов из спиртов и аминов, а вода может гидролизовать ангидриды до соответствующих кислот. Как и галогенангидриды, ангидриды также могут реагировать с углеродными нуклеофилами с образованием кетонов и/или третичных спиртов, а также могут участвовать как в ацилировании Фриделя-Крафтса, так и в синтезе кетонов Вайнреба. [8] Однако, в отличие от галогенангидридов, ангидриды не реагируют с реагентами Гилмана. [2]
Реакционную способность ангидридов можно повысить, используя каталитическое количество N,N-диметиламинопиридина или ДМАП. пиридин , который действует по аналогичному механизму. Для этой цели также можно использовать [5]

Сначала ДМАП ( 2 ) атакует ангидрид ( 1 ) с образованием тетраэдрического промежуточного продукта, который разрушается, удаляя карбоксилат-ион и образуя амид 3 . Этот промежуточный амид более активируется в отношении нуклеофильной атаки, чем исходный ангидрид, поскольку диметиламинопиридин является лучшей уходящей группой, чем карбоксилат. На последнем этапе нуклеофил (Nuc) атакует 3 , образуя еще один тетраэдрический промежуточный продукт. Когда это промежуточное соединение распадается с образованием продукта 4 , пиридиновая группа удаляется и его ароматичность восстанавливается – мощная движущая сила и причина, по которой пиридиновое соединение является лучшей уходящей группой, чем карбоксилат-ион.
Эфиры
[ редактировать ]Эфиры менее реакционноспособны, чем галогенангидриды и ангидриды. Как и более реакционноспособные ацильные производные, они могут реагировать с аммиаком , а также первичными и вторичными аминами с образованием амидов, хотя этот тип реакции используется не часто, поскольку галогенангидриды дают лучшие выходы. Эфиры могут быть преобразованы в другие сложные эфиры в процессе, известном как переэтерификация . Переэтерификация может катализироваться кислотами или основаниями и включает реакцию сложного эфира со спиртом. К сожалению, поскольку уходящая группа также представляет собой спирт, прямая и обратная реакции часто протекают с одинаковой скоростью. Использование большого избытка реагирующего спирта или удаление спирта уходящей группы (например, путем перегонки ) приведет к завершению прямой реакции в соответствии с принципом Ле Шателье . [9]
Кислотно-катализируемый гидролиз сложных эфиров также является равновесным процессом, по существу обратным реакции этерификации Фишера . Поскольку спирт (действующий как уходящая группа) и вода (действующая как нуклеофил) имеют одинаковые значения p K a , прямая и обратная реакции конкурируют друг с другом. Как и при переэтерификации, использование большого избытка реагента (воды) или удаление одного из продуктов (спирта) может способствовать прямой реакции.
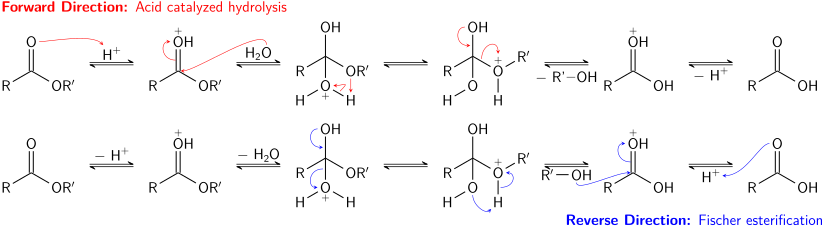
Основной гидролиз сложных эфиров, известный как омыление , не является равновесным процессом; В реакции расходуется полный эквивалент основания, в результате чего образуется один эквивалент спирта и один эквивалент карбоксилатной соли. Омыление эфиров жирных кислот — промышленно важный процесс, используемый при производстве мыла. [9]
Эфиры могут вступать в различные реакции с углеродными нуклеофилами. Как и галогенангидриды и ангириды, они реагируют с избытком реактива Гриньяра с образованием третичных спиртов. Эфиры также легко реагируют с енолятами . При конденсации Кляйзена енолят одного сложного эфира ( 1 ) атакует карбонильную группу другого сложного эфира ( 2 ) с образованием тетраэдрического промежуточного соединения 3 . Промежуточное соединение разрушается, вытесняя алкоксид (R'O − ) и получение β-кетоэфира 4 .

Возможны также скрещенные конденсации Кляйзена, в которых енолятом и нуклеофилом являются разные эфиры. Внутримолекулярную или циклизацией Дикмана, поскольку ее конденсацию Кляйзена называют конденсацией Дикмана можно использовать для образования колец. Эфиры также могут подвергаться конденсации с енолятами кетонов и альдегидов с образованием β-дикарбонильных соединений. [10] Конкретным примером этого является перегруппировка Бейкера-Венкатарамана , при которой ароматический орто -ацилоксикетон подвергается внутримолекулярному нуклеофильному ациловому замещению и последующей перегруппировке с образованием ароматического β-дикетона. [11] Перегруппировка Чана является еще одним примером перегруппировки, возникающей в результате внутримолекулярной реакции нуклеофильного ацильного замещения.
Амиды
[ редактировать ]Из-за своей низкой реакционной способности амиды не участвуют в таком количестве реакций нуклеофильного замещения, как другие ацильные производные. Амиды устойчивы к воде и примерно в 100 раз более устойчивы к гидролизу, чем сложные эфиры. [3] Однако амиды могут гидролизоваться до карбоновых кислот в присутствии кислоты или основания. Стабильность амидных связей имеет биологическое значение, поскольку аминокислоты , входящие в состав белков, связаны амидными связями. Амидные связи достаточно устойчивы к гидролизу, чтобы сохранять структуру белка в водной среде, но достаточно чувствительны, чтобы при необходимости их можно было разорвать. [3]
Первичные и вторичные амиды плохо реагируют с углеродными нуклеофилами. Реактивы Гриньяра и литийорганические соединения будут действовать как основания, а не нуклеофилы, и просто депротонируют амид. Третичные амиды не сталкиваются с этой проблемой и реагируют с углеродными нуклеофилами с образованием кетонов ; амид - анион (NR 2 − ) является очень сильным основанием и, следовательно, очень плохой уходящей группой, поэтому нуклеофильная атака происходит только один раз. При взаимодействии с углеродными нуклеофилами N , N -диметилформамид (ДМФ) может быть использован для введения формильной группы. [12]

Здесь фениллитий 1 атакует карбонильную группу ДМФ 2 , образуя тетраэдрический интермедиат 3 . Поскольку диметиламид-анион является плохой уходящей группой, промежуточное соединение не разрушается и другого нуклеофильного присоединения не происходит. При кислой обработке алкоксид протонируется с образованием 4 , затем протонируется амин с образованием 5 . Отщепление нейтральной молекулы диметиламина и потеря протона дают бензальдегид 6 .
Карбоновые кислоты
[ редактировать ]Карбоновые кислоты не особенно реакционноспособны в отношении нуклеофильного замещения, хотя их можно превратить в другие ацильные производные. Преобразование карбоновой кислоты в амид возможно, но не просто. Вместо того, чтобы действовать как нуклеофил, амин будет реагировать как основание в присутствии карбоновой кислоты с образованием карбоксилатной соли аммония. Нагревание соли до температуры выше 100 °C приведет к удалению воды и образованию амида. Этот метод синтеза амидов имеет промышленное значение, а также имеет лабораторное применение. [13] В присутствии сильнокислотного катализатора карбоновые кислоты могут конденсироваться с образованием ангидридов кислот. Однако в результате конденсации образуется вода, которая может гидролизовать ангидрид обратно до исходных карбоновых кислот. Таким образом, образование ангидрида путем конденсации является равновесным процессом.
В условиях кислотного катализа карбоновые кислоты вступают в реакцию со спиртами с образованием сложных эфиров посредством реакции этерификации Фишера , которая также является равновесным процессом. Альтернативно, диазометан для превращения кислоты в сложный эфир можно использовать . Хотя реакции этерификации с диазометаном часто дают количественные выходы, диазометан полезен только для образования метиловых эфиров. [13]
Тионилхлорид можно использовать для преобразования карбоновых кислот в соответствующие ацилхлориды. Сначала карбоновая кислота 1 атакует тионилхлорид, и хлорид-ион уходит. Образующийся ион оксония 2 активируется в направлении нуклеофильной атаки и имеет хорошую уходящую группу, что отличает его от обычной карбоновой кислоты. На следующем этапе 2 подвергается атаке хлорид-иона с образованием тетраэдрического промежуточного соединения 3 – хлорсульфита. Тетраэдрический интермедиат разрушается с потерей диоксида серы и иона хлорида, образуя протонированный ацилхлорид 4 . Хлорид-ион может отобрать протон карбонильной группы, давая ацилхлорид 5 с потерей HCl .

Хлорид фосфора(III) (PCl 3 ) и хлорид фосфора(V) (PCl 5 ) также преобразуют карбоновые кислоты в хлорангидриды по аналогичному механизму. Один эквивалент PCl 3 может реагировать с тремя эквивалентами кислоты, образуя один эквивалент H 3 PO 3 или фосфорной кислоты в дополнение к желаемому хлорангидриду. PCl 5 реагирует с карбоновыми кислотами в соотношении 1:1 с образованием оксихлорида фосфора(V) (POCl 3 ) и хлористого водорода (HCl) в качестве побочных продуктов.
Карбоновые кислоты реагируют с реактивами Гриньяра и литийорганическими соединениями с образованием кетонов. Первый эквивалент нуклеофила действует как основание и депротонирует кислоту. Второй эквивалент атакует карбонильную группу с образованием геминального алкоксидного дианиона, который при обработке протонируется с образованием гидрата кетона. Поскольку большинство гидратов кетонов нестабильны по отношению к соответствующим кетонам, равновесие между ними сильно смещается в пользу кетона. Например, константа равновесия образования гидрата ацетона из ацетона составляет всего 0,002. Карбоксильная группа – самая кислая в органических соединениях. [14]
См. также
[ редактировать ]- Нуклеофильное алифатическое замещение
- Нуклеофильное ароматическое замещение
- Нуклеофильная абстракция
Ссылки
[ редактировать ]- ^ Уэйд 2010, стр. 996–997.
- ^ Jump up to: а б с Макмерри, Джон (1996). Органическая химия (4-е изд.). Пасифик Гроув, Калифорния: Издательская компания Brooks/Cole. стр. 820–821 . ISBN 0534238327 .
- ^ Jump up to: а б с д Кэри, Фрэнсис А. (2006). Органическая химия (6-е изд.). Нью-Йорк: МакГроу-Хилл. стр. 866–868 . ISBN 0072828374 .
- ^ Jump up to: а б с Уэйд 2010, стр. 998–999.
- ^ Jump up to: а б Курти, Ласло; Барбара Чако (2005). Стратегическое применение названных реакций в органическом синтезе . Лондон: Elsevier Academic Press. п. 398. ИСБН 0124297854 .
- ^ Макмерри 1996, стр. 826–827.
- ^ Курти и Чако 2005, с. 478.
- ^ Jump up to: а б Курти и Чако 2005, с. 176.
- ^ Jump up to: а б Уэйд 2010, стр. 1005–1009.
- ^ Кэри 2006, стр. 919–924.
- ^ Курти и Чако 2005, с. 30.
- ^ Алан Р. Катрицки ; Мет-Кон, Отто; Чарльз Рис , ред. (1995). Комплексные преобразования органических функциональных групп . Том. 3 (1-е изд.). Оксфорд: Пергамон Пресс. п. 90 . ISBN 0080423248 .
- ^ Jump up to: а б Уэйд 2010, стр. 964–965.
- ^ Уэйд 2010, с. 838.
Внешние ссылки
[ редактировать ]- Реакция уксусного ангидрида с ацетоном в органическом синтезе . Том. 3, с. 16; Том. 20, с. 6 Статья