Енолат
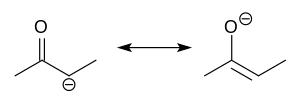
В органической химии еноляты — это органические анионы, в результате депротонирования карбонила образующиеся ( RR'C=O ) соединения. Редко выделяемые, они широко используются в качестве реагентов при синтезе органических соединений . [1] [2] [3] [4]
Склеивание и структура
[ редактировать ]
Енолят-анионы электронно связаны с аллильными анионами. Анионный заряд делокализован по кислороду и двум углеродным узлам. Таким образом, они имеют характер как алкоксида , так и карбаниона . [5]
Хотя их часто изображают как простые соли, на самом деле они имеют сложную структуру, часто состоящую из агрегатов. [6]

Подготовка
[ редактировать ]Депротонирование енолизируемых кетонов, ароматических спиртов, альдегидов и сложных эфиров дает еноляты. [8] [9] В случае сильных оснований депротонирование носит количественный характер. Обычно еноляты получают при использовании диизопропиламида лития (LDA). [10]
Часто, как и в обычных конденсациях Кляйзена , реакциях Манниха и альдольных конденсациях , еноляты образуются в низких концентрациях с алкоксидными основаниями. В таких условиях они существуют в небольших концентрациях, но все же вступают в реакции с электрофилами. На поведение енолятов влияют многие факторы, особенно растворитель, добавки (например, диамины) и противокатион (Li + против На + , и т. д.). Для несимметричных кетонов существуют методы контроля региохимии депротонирования. [11]

Депротонирование углеродных кислот может протекать как при кинетическом, так и при термодинамическом контроле реакции . Например, в случае фенилацетона депротонирование может привести к образованию двух разных енолятов. Было показано, что LDA депротонирует метильную группу, что является кинетическим ходом депротонирования. Для обеспечения получения кинетического продукта используют небольшой избыток (1,1 экв.) диизопропиламида лития и добавляют кетон к основанию при -78 °С. Поскольку кетон быстро и количественно превращается в енолят, а основание всегда присутствует в избытке, кетон не может действовать как переносчик протонов, катализируя постепенное образование термодинамического продукта. Более слабое основание, такое как алкоксид , который обратимо депротонирует субстрат, дает более термодинамически стабильный бензиленолят.
Еноляты могут улавливаться путем ацилирования и силилирования , которые происходят по кислороду. Эфиры силилинола являются обычными реагентами в органическом синтезе, о чем свидетельствует альдольная реакция Мукаямы : [13]

Роль кислот Льюиса в образовании енолятов
[ редактировать ]Помимо использования сильных оснований, еноляты можно получить с использованием кислоты Льюиса и слабого основания («мягкие условия»):

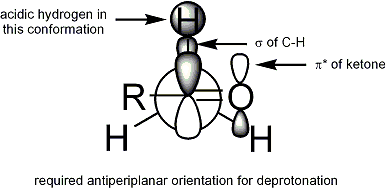
Для того чтобы депротонирование произошло , стереоэлектронное требование состоит в том, чтобы сигма-связь альфа-СН могла перекрываться с pi*-орбиталью карбонила :
Геометрия
[ редактировать ]Были проведены обширные исследования образования енолятов. В большинстве случаев можно создать желаемую енолятную геометрию: [14]
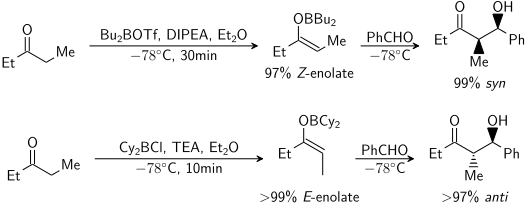
Что касается кетонов, то в большинстве условий енолизации образуются Z. еноляты Для сложных эфиров большинство условий енолизации дают E. еноляты добавление HMPA Известно, что обращает вспять стереоселективность депротонирования.

Стереоселективное образование енолятов было объяснено ирландской моделью . [15] [16] [17] [18] хотя его достоверность несколько сомнительна. В большинстве случаев неизвестно, какие промежуточные соединения (если таковые имеются) являются мономерными или олигомерными по своей природе; тем не менее, модель Ирландии остается полезным инструментом для понимания енолятов.
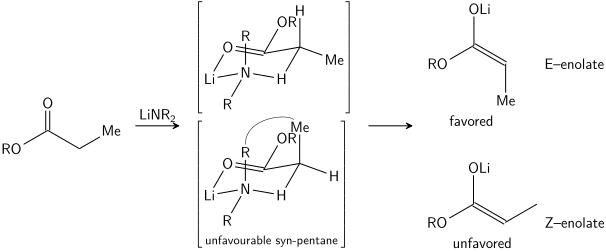
В модели Ирландии предполагается, что депротонирование протекает по шестичленному или циклическому механизму. [19] мономерное переходное состояние . Больший из двух заместителей электрофила (в приведенном выше случае метил больше протона) принимает экваториальное расположение в предпочтительном переходном состоянии, что приводит к предпочтению енолятов E. Модель явно терпит неудачу во многих случаях; например, если смесь растворителей заменить ТГФ на 23% ГМПА-ТГФ (как показано выше), геометрия енолята изменится на противоположную, что несовместимо с этой моделью и ее циклическим переходным состоянием.
Региохимия образования енолята
[ редактировать ]Если несимметричный кетон подвергается воздействию основания, он может образовать два региоизомерных енолята (игнорируя геометрию енолята). Например:
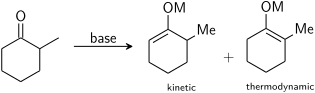
Трехзамещенный енолят считается кинетическим енолятом, а тетразамещенный енолят считается термодинамическим енолятом. Альфа-водород, депротонированный с образованием кинетического енолята, менее затруднен и, следовательно, депротонируется быстрее. В общем, тетразамещенные олефины более стабильны, чем тризамещенные олефины, благодаря гиперконъюгативной стабилизации. На соотношение енолятных региоизомеров сильно влияет выбор основания. В приведенном выше примере кинетический контроль может быть установлен с помощью LDA при -78 ° C, что дает селективность кинетического: термодинамического енолята 99:1, тогда как термодинамический контроль может быть установлен с помощью трифенилметиллития при комнатной температуре , что дает селективность 10:90.
В целом, кинетическим енолятам благоприятствуют низкие температуры, условия, которые обеспечивают относительно ионную связь металл-кислород, и быстрое депротонирование с использованием небольшого избытка сильного, стерически затрудненного основания. Большое основание депротонирует только более доступный водород, а низкие температуры и избыток основания помогают избежать уравновешивания с более стабильным альтернативным енолятом после первоначального образования енолята. Термодинамическим енолятам благоприятствует более длительное время установления равновесия при более высоких температурах, условия, которые обеспечивают относительно ковалентную связь металл-кислород, и использование небольшого субстехиометрического количества сильного основания. При использовании недостаточного количества основания для депротонирования всех карбонильных молекул еноляты и карбонилы могут обмениваться протонами друг с другом и уравновешиваться до своего более стабильного изомера. Использование различных металлов и растворителей может обеспечить контроль над количеством ионного характера в связи металл-кислород.
Реакции
[ редактировать ]Будучи мощными нуклеофилами, еноляты легко реагируют с различными электрофилами. Эти реакции генерируют новые связи CC и часто новые стереоцентры. На стереоселективность и региоселективность влияют добавки, растворитель, противоионы и т. д. Одним из важных классов электрофилов являются алкилгалогениды, и в этом случае возникает классическая проблема: O-алкилирование против C-алкилирования . Контроль этой избирательности привлек много внимания. Отрицательный заряд в енолятах сосредоточен на кислороде, но этот центр также сильно сольватирован, что приводит к C-алкилированию. [20]
Другими важными электрофилами являются альдегиды/кетоны и акцепторы Михаэля . [21]

Sample aldol reaction with lithium enolate

Синтез енонов с использованием региоспецифического образования енолятов и скрытой функциональности.
[ редактировать ]

Региоспецифическое образование представляет собой контролируемое образование енолята путем специфического депротонирования одного из α-углеродов исходной молекулы кетона. Это обеспечивает одну из наиболее понятных синтетических стратегий для внесения химической сложности в натуральные продукты и общий синтез . Ярким примером его использования является общий синтез прогестерона, показанный на рисунке «Образование региоспецифического енолята при полном синтезе прогестерона».
Когда кетоны обрабатываются основанием , еноляты могут образовываться путем депротонирования любого α-углерода. Селективность определяется как стерическими , так и электронными эффектами на α-углероды, а также точным используемым основанием (пример этого см. на рисунке «Маскированная функциональность» для образования региоспецифического енолята). Образование енолята будет термодинамически предпочтительным при наиболее кислом протоне, что зависит от электронной стабилизации образующегося аниона . Однако селективность можно обратить вспять, стерически препятствуя термодинамическому продукту и, следовательно, кинетически благоприятствуя депротонированию в другом α-углеродном центре. Традиционные методы региоселективного образования енолятов используют либо электронные активирующие группы (например, альдегиды ), либо стерические блокирующие группы (например, 1,2-этандитиол- защищенный кетон).
Енон также может служить предшественником региоспецифического образования енолята, здесь енон представляет собой «замаскированную функциональность» енолята. Этот процесс впервые описал Гилберт Сторк. [22] который наиболее известен своим вкладом в изучение методов селективного образования енолятов в органическом синтезе . Реакция енона с металлическим литием приводит к образованию енолята у α-углерода енона. Енолятный продукт можно либо улавливать, либо алкилировать. Используя «замаскированную функциональность», можно производить еноляты, недоступные традиционными методами.
Подход «замаскированной функциональности» к образованию региоспецифических енолятов широко используется при полном синтезе натуральных продуктов. Например, при общем синтезе стероидного гормона прогестерона , [23] Сторк и его коллеги использовали «замаскированную функциональность» для стереоспецифического построения одного из четвертичных атомов углерода в молекуле.
Избегайте енолятов
[ редактировать ]Азаеноляты (также известные как имин-анионы, енамиды, металлизированные основания Шиффа и металлоенамины) представляют собой азотистые аналоги енолятов. [24] Когда имины обрабатываются сильными основаниями, такими как LDA , образуются высоконуклеофильные азаеноляты.
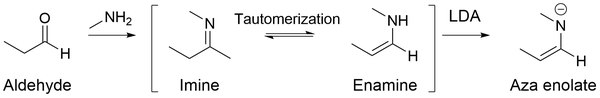
Основное преимущество использования азаенолятов заключается в том, что они не подвергаются ) в основном или самоконденсации (т.е. альдольной реакции альдегидов нейтральном растворе, а скорее способствуют алкилированию по альфа-углероду. [25] Это происходит главным образом потому, что имины содержат двойные связи углерод-азот в отличие от альдегидов, которые содержат двойные связи кислород-углерод. Поскольку кислород более электроотрицательен, чем азот, он отбирает большую электронную плотность у карбонильного углерода, создавая больший частично положительный заряд на углероде. Следовательно, имея более электрофильный углерод, альдегиды обеспечивают лучшее нуклеофильное присоединение к углероду по двойной связи углерод-кислород.
С другой стороны, имин имеет меньше электроотрицательного азота, что индуцирует более слабый частично положительный заряд на карбонильном углероде. В результате, хотя имины все еще могут реагировать с литийорганическими соединениями, они не реагируют с другими нуклеофилами (включая азаеноляты) с образованием нуклеофильных присоединений . [26]
Вместо этого азаеноляты реагируют аналогично енолятам, образуя SN2- алкилированные продукты. [25] В результате конъюгации неподеленной пары азота β-углерод становится нуклеофильным участком, позволяя азаенолятам подвергаться реакциям алкилирования. [27] Таким образом, азаеноляты могут реагировать с многочисленными электрофилами, такими как эпоксиды и алкилгалогениды, с образованием новой углерод-углеродной связи на β-углероде. [24]
Ниже показаны два потенциальных механизма реакции:

Поскольку эпоксид представляет собой трехчленную кольцевую молекулу, он имеет высокую степень деформации кольца . Хотя атомы углерода в кольцевой системе являются тетраэдрическими , предпочтительно 109,5 градусов между каждым атомом, эпоксид деформирует углы кольца до 60 градусов. Чтобы противостоять этому эффекту, нуклеофильные азаеноляты легко реагируют с эпоксидами, уменьшая напряжения их колец.

Помимо реакции с эпоксидами, азаеноляты могут также реагировать с алкилгалогенидами (или аллилгалогенидами, как показано выше) с образованием новой сигма-связи углерод-углерод . Эта реакция является одним из ключевых этапов синтеза феромона мужской агрессии Oulema melanopus. [29] Азаенолят образуется в результате реакции LDA с пивальдегидом, который затем реагирует с алкилгалогенидом с образованием промежуточного продукта Oulema melanopus.
Еноляты аза также могут образовываться с помощью реактивов Гриньяра и вступать в реакцию с другими мягкими электрофилами, включая рецепторы Михаэля . [24]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Штольц, Дэниел; Казмайер, Ули (2010). «Еноляты металлов как синтоны в органической химии». Химия функциональных групп PATai . дои : 10.1002/9780470682531.pat0423 . ISBN 978-0-470-68253-1 .
- ^ Харт, Дэвид Дж.; Ха, Док Чан (1989). «Путь конденсации енолята-имина сложного эфира с образованием бета-лактамов». Химические обзоры . 89 (7): 1447–1465. дои : 10.1021/cr00097a003 .
- ^ Ву, Джордж; Хуан, Миншэн (2006). «Литийорганические реагенты в фармацевтических асимметричных процессах». Химические обзоры . 106 (7): 2596–2616. дои : 10.1021/cr040694k . ПМИД 16836294 .
- ^ Курти, Клаудио; Баттистини, Люсия; Сартори, Андреа; Занарди, Франка (2020). «Новые разработки принципа винилологии применительно к π-расширенным донорским системам енолятного типа» . Химические обзоры . 120 (5): 2448–2612. doi : 10.1021/acs.chemrev.9b00481 . ПМЦ 7993750 . ПМИД 32040305 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Эноляты ». doi : 10.1351/goldbook.E02123
- ^ Райх, Ханс Дж. (2013). «Роль литийорганических агрегатов и смешанных агрегатов в литийорганических механизмах». Химические обзоры . 113 (9): 7130–7178. дои : 10.1021/cr400187u . ПМИД 23941648 .
- ^ Николс, Майкл А.; Лепоса, Кристина М.; Хантер, Аллен Д.; Целлер, Матиас (2007). «Кристаллические структуры гексамерных и димерных комплексов литиоизобутирофенона». Журнал химической кристаллографии . 37 (12): 825–829. дои : 10.1007/s10870-007-9255-0 . S2CID 97183362 .
- ^ Смит, Майкл Б.; Марч, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Манфред Браун (2015). Современная химия енолятов: от получения к применению в асимметричном синтезе . Вили-ВЧ. дои : 10.1002/9783527671069 . ISBN 978-3-527-67106-9 .
- ^ Кристин Ведлер; Ганс Шик (1998). «Синтез β-лактонов путем альдолизации кетонов енолятами фениловых эфиров: 3,3-диметил-1-оксаспиро[3,5]нонан-2-он». Орг. Синтез . 75 : 116. дои : 10.15227/orgsyn.075.0116 .
- ^ Галл, Мартин; Хаус, Герберт О. (1972). «Образование и алкилирование специфических енолят-анионов из несимметричного кетона: 2-бензил-2-метилциклогексанона и 2-бензил-6-метилциклогексанона». Орг. Синтез . 52 : 39. дои : 10.15227/orgsyn.052.0039 .
- ^ Конг, Цзяньше; Мэн, Тао; Тинг, Полина; Вонг, Джесси (2010). «Получение этил-1-бензил-4-фторпиперидин-4-карбоксилата» . Органические синтезы . 87 : 137. дои : 10.15227/orgsyn.087.0137 .
- ^ Мукаяма, Теруаки; Кобаяши, Шу (1994). «Еноляты олова (II) в реакциях Альдола, Михаэля и родственных им». Органические реакции . стр. 1–103. дои : 10.1002/0471264180.или046.01 . ISBN 0-471-26418-0 .
- ^ Браун, ХК ; Дхар, РК; Бакши, РК; Пандиараджан, ПК; Сингарам, Б. (1989). «Основное влияние уходящей группы в хлоридах и трифлатах диалкилбора на контроль стереоспецифического превращения кетонов в E- или Z-енолборинаты». Журнал Американского химического общества . 111 (9): 3441–3442. дои : 10.1021/ja00191a058 .
- ^ Ирландия, RE; Уиллард, АК (1975). «Стереоселективное поколение енолятов сложных эфиров». Буквы тетраэдра . 16 (46): 3975–3978. дои : 10.1016/S0040-4039(00)91213-9 .
- ^ Нарула, А.С. (1981). «Анализ взаимодействий диастереомерных переходных состояний для кинетического депротонирования ациклических карбонильных производных диизопропиламидом лития». Буквы тетраэдра . 22 (41): 4119–4122. дои : 10.1016/S0040-4039(01)82081-5 .
- ^ Ирландия, RE; Випф, П; Армстронг, доктор юридических наук (1991). «Стереохимический контроль перегруппировки Кляйзена в еноляте сложного эфира. 1. Стереоселективность в образовании силилкетенацеталя». Журнал органической химии . 56 (2): 650–657. дои : 10.1021/jo00002a030 .
- ^ Се, Л; Айзенбергер, К.М.; Хелд, Г; Даль, Л.М. (октябрь 1997 г.). «Высокостереоселективное кинетическое образование енолята: стерические и электронные эффекты». Журнал органической химии . 62 (21): 7516–7519. дои : 10.1021/jo971260a . ПМИД 11671880 .
- ^ Направленный синтез альдолов - образование E-енолята и Z-енолята.
- ^ Смит, Майкл Б.; Марч, Джерри (2007), Продвинутая органическая химия: реакции, механизмы и структура (6-е изд.), Нью-Йорк: Wiley-Interscience, стр. 551, ISBN 978-0-471-72091-1
- ^ Зеебах, Дитер (1988). «Структура и реакционная способность енолятов лития. От пинаколона к селективному C-алкилированию пептидов. Трудности и возможности, открываемые сложными структурами». Angewandte Chemie International Edition на английском языке . 27 (12): 1624–1654. дои : 10.1002/anie.198816241 .
- ^ Аист, Г.; Сингх, Дж., Дж. Ам. хим. Соц. 1974, 96, 6181.
- ^ Аист, Г.; Макмерри, Дж. Э., Дж. Ам. хим. Соц. 1967, 89, 5464.
- ^ Jump up to: а б с Аслам, О. (28 сентября 2012 г.). Разработка каталитических азаенолятных реакций (докторская диссертация). UCL (Университетский колледж Лондона).
- ^ Jump up to: а б Клейден, Джонатан (2012). Органическая химия (2-е изд.). Оксфорд: Издательство Оксфордского университета. стр. 465, 593–594. ISBN 978-0-19-927029-3 .
- ^ Крэнвелл, Филиппа. «Энамины/аза-еноляты – Механизм Мордор» . сайты.google.com . Архивировано из оригинала 3 сентября 2021 г. Проверено 28 ноября 2020 г.
- ^ Кэри, Фрэнсис А. (2007). Передовая органическая химия. Часть B, Реакции и синтез (5-е изд.). Нью-Йорк, штат Нью-Йорк: Спрингер. стр. 46–47. ISBN 978-0-387-68350-8 .
- ^ Худрлик, Пол Ф.; Ван, Чунг-Нан (октябрь 1975 г.). «Реакции оксетана с иминными солями, полученными из циклогексанона». Журнал органической химии . 40 (20): 2963–2965. дои : 10.1021/jo00908a027 .
- ^ Jump up to: а б Шевалле, Алиса; Ферезу, Жан-Пьер (2012). «Образование азаенолятов из вторичных аминов в одном котле и конденсация до сложных эфиров и алкилбромидов». Тетраэдр . 68 (29): 5882–5889. дои : 10.1016/j.tet.2012.04.105 .
| Базы данных органов управления : Национальные |
|---|