Физическая органическая химия
В этой статье есть несколько проблем. Пожалуйста, помогите улучшить его или обсудите эти проблемы на странице обсуждения . ( Узнайте, как и когда удалять эти шаблонные сообщения ) |
Физическая органическая химия , термин, придуманный Луи Хэмметом в 1940 году, относится к дисциплине органической химии , которая фокусируется на взаимосвязи между химическими структурами и реакционной способностью , в частности, на применении экспериментальных инструментов физической химии для изучения органических молекул . Конкретные фокусы исследования включают скорость , органических реакций относительную химическую стабильность исходных материалов, реакционноспособных промежуточных продуктов , переходных состояний и продуктов химических реакций , а также нековалентные аспекты сольватации и молекулярных взаимодействий , которые влияют на химическую реакционную способность. Такие исследования обеспечивают теоретическую и практическую основу для понимания того, как изменения структуры в растворе или твердом состоянии влияют на механизм и скорость каждой органической реакции интересующей .
Приложение [ править ]
Физические химики-органики используют теоретические и экспериментальные подходы для понимания этих фундаментальных проблем органической химии , включая классические и статистические термодинамические расчеты, квантово-механическую теорию и вычислительную химию , а также экспериментальную спектроскопию (например, ЯМР ), спектрометрию (например, МС ), и кристаллографический подходы. Таким образом, эта область находит применение в широком спектре более специализированных областей, включая электро- и фотохимию , химию полимеров и супрамолекулярную химию , биоорганическую химию , энзимологию и химическую биологию , а также для коммерческих предприятий, занимающихся химией процессов , химической инженерией , материаловедением. и нанотехнологии , и фармакология в разработке лекарств .
Область применения [ править ]
Физическая органическая химия — это изучение взаимосвязи между структурой и реакционной способностью органических молекул . В частности, физическая органическая химия применяет экспериментальные инструменты физической химии для изучения структуры органических молекул и обеспечивает теоретическую основу, которая интерпретирует, как структура влияет как на механизмы , так и на скорость органических реакций . Ее можно рассматривать как подполе, которое соединяет органическую химию с физической химией .
Физические химики-органики используют как экспериментальные, так и теоретические дисциплины, такие как спектроскопия , спектрометрия , кристаллография , вычислительная химия и квантовая теория , для изучения как скоростей , органических реакций так и относительной химической стабильности исходных материалов, переходных состояний и продуктов. [1] [ нужна страница ] Химики в этой области работают над пониманием физических основ современной органической химии , и поэтому физическая органическая химия находит применение в специализированных областях, включая химию полимеров , супрамолекулярную химию , электрохимию и фотохимию . [1] [ нужна страница ]
История [ править ]
Сам термин «физическая органическая химия» был придуман Луи Хэмметом в 1940 году, когда он использовал эту фразу в качестве названия для своего учебника. [2]
Химическая структура и термодинамика [ править ]
Термохимия [ править ]
Химики-органики используют инструменты термодинамики для изучения связей , стабильности и энергетики химических систем. Сюда входят эксперименты по измерению или определению энтальпии (Δ H ), энтропии (Δ S ) и свободной энергии Гиббса (Δ G ) реакции, превращения или изомеризации. Для расчета этих значений химики могут использовать различные химические и математические анализы, такие как график Ван-т-Гоффа .
Эмпирические константы, такие как энергия диссоциации связи , стандартная теплота образования (Δ f H °) и теплота сгорания (Δ c H °), используются для прогнозирования стабильности молекул и изменения энтальпии (Δ H ) в ходе реакции. Для сложных молекул значение Δ f H ° может быть недоступно, но его можно оценить, используя молекулярные фрагменты с известной теплотой образования . Этот тип анализа часто называют теорией приращения группы Бенсона в честь химика Сидни Бенсона, который всю карьеру разрабатывал эту концепцию. [1] [ нужна страница ] [3] [4]
Термохимия реакционноспособных промежуточных продуктов — карбокатионов , карбанионов и радикалов — представляет интерес и для физико-химиков-органиков. Данные о групповом приращении доступны для радикальных систем. [1] [ нужна страница ] Стабильность карбокатионов и карбанионов можно оценить, используя сродство к гидрид-иону и pK a значения соответственно. [1] [ нужна страница ]
Конформационный анализ [ править ]
Одним из основных методов оценки химической стабильности и энергетики является конформационный анализ . Физические химики-органики используют конформационный анализ для оценки различных типов штаммов , присутствующих в молекуле, и прогнозирования продуктов реакции. [5] [ нужна страница ] Деформацию можно обнаружить как в ациклических, так и в циклических молекулах, проявляясь в различных системах, таких как деформация кручения , аллильная деформация , кольцевая деформация и синпентановая деформация . [1] [ нужна страница ] Значения A обеспечивают количественную основу для прогнозирования конформации замещенного циклогексана , важного класса циклических органических соединений, реакционная способность которых в значительной степени определяется конформационными эффектами. Значение A представляет собой разницу в свободной энергии Гиббса между аксиальной и экваториальной формами замещенного циклогексана, и путем сложения значений A различных заместителей можно количественно предсказать предпочтительную конформацию производного циклогексана.
Помимо молекулярной стабильности, конформационный анализ используется для прогнозирования продуктов реакции. Одним из часто цитируемых примеров использования конформационного анализа является реакция бимолекулярного элиминирования (E2). Эта реакция протекает наиболее легко, когда нуклеофил атакует молекулу, антиперипланарную по отношению к уходящей группе. Молекулярно -орбитальный анализ этого явления позволяет предположить, что эта конформация обеспечивает наилучшее перекрытие между электронами на связывающей орбитали RH σ , которая подвергается нуклеофильной атаке, и пустой разрыхляющей орбиталью σ* связи RX, которая разрывается. [6] [ нужна страница ] Используя этот эффект, конформационный анализ можно использовать для создания молекул, обладающих повышенной реакционной способностью.
Физические процессы, которые приводят к возникновению барьеров вращения связей , сложны, и эти барьеры тщательно изучались экспериментальными и теоретическими методами. [7] [8] [9] В ряде недавних статей исследовалось преобладание стерического , электростатического и гиперконъюгативного вклада во вращательные барьеры в этане , бутане и других замещенных молекулах. [10]
Нековалентные взаимодействия [ править ]

Химики используют изучение внутримолекулярных и межмолекулярных нековалентных связей/взаимодействий в молекулах для оценки реакционной способности. Такие взаимодействия включают, помимо прочего, водородную связь , электростатические взаимодействия между заряженными молекулами, диполь-дипольные взаимодействия , полярные π- и катион-π- взаимодействия, π-стекинг , донорно-акцепторную химию и галогенную связь . Кроме того, гидрофобный эффект — ассоциация органических соединений в воде — представляет собой электростатическое нековалентное взаимодействие , представляющее интерес для химиков. Точная физическая природа гидрофобного эффекта обусловлена многими сложными взаимодействиями , но считается, что он является наиболее важным компонентом биомолекулярного распознавания в воде. [1] [ нужна страница ] Например, исследователи выяснили структурную основу распознавания фолиевой кислоты белками-рецепторами фолиевой кислоты. [11] Сильное взаимодействие между фолиевой кислотой и фолатным рецептором объясняется как водородными связями , так и гидрофобными взаимодействиями . Исследование нековалентных взаимодействий также используется для изучения связывания и кооперативности в супрамолекулярных ансамблях и макроциклических соединениях, таких как краун-эфиры и криптанды , которые могут выступать в качестве хозяев для гостевых молекул.
Кислотно-основная химия [ править ]

Свойства кислот и оснований имеют отношение к физической органической химии. Химики-органики в первую очередь интересуются Бренстеда-Лоури кислотами/основаниями как донорами/акцепторами протонов, а кислотами/основаниями Льюиса как акцепторами/донорами электронов в органических реакциях. Химики используют ряд факторов, разработанных на основе физической химии — электроотрицательность / индукция , прочность связей , резонанс , гибридизация , ароматичность и сольватация — для прогнозирования относительной кислотности и основности.
Принцип « жесткая/мягкая кислота/основание» используется для прогнозирования молекулярных взаимодействий и направления реакции. В общем, предпочтительны взаимодействия между молекулами одного типа. То есть жёсткие кислоты будут ассоциироваться с твёрдыми основаниями, а мягкие кислоты с мягкими основаниями. Представления о жестких кислотах и основаниях часто используются при синтезе неорганических координационных комплексов .
Кинетика [ править ]
Физические химики-органики используют математические основы химической кинетики для изучения скоростей реакций и механизмов реакций. В отличие от термодинамики, которая занимается вопросами относительной стабильности продуктов и реагентов (ΔG ° ) и их равновесных концентраций, при изучении кинетики основное внимание уделяется свободной энергии активации ( ΔG ‡ ) — разница в свободной энергии между структурой реагента и структурой переходного состояния — реакции и, следовательно, позволяет химику изучать процесс уравновешивания . [1] [ нужна страница ] математически выведенные формализмы, такие как постулат Хаммонда , принцип Кертина-Хэммета и теория микроскопической обратимости часто применяются К органической химии . Химики также использовали принцип термодинамического и кинетического контроля , чтобы влиять на продукты реакции.
Законы о ставках [ править ]
Изучение химической кинетики используется для определения закона скорости реакции. Закон скорости обеспечивает количественную связь между скоростью химической реакции и концентрацией или давлением присутствующих химических веществ. [12] [ нужна страница ] Законы скорости должны определяться экспериментальными измерениями и, как правило, не могут быть объяснены из химического уравнения . Экспериментально определенный закон скорости относится к стехиометрии структуры переходного состояния относительно структуры основного состояния. Определение закона скорости исторически осуществлялось путем мониторинга концентрации реагента во время реакции посредством гравиметрического анализа , но сегодня это делается почти исключительно с помощью быстрых и однозначных спектроскопических методов. В большинстве случаев определение уравнений скорости упрощается добавлением большого избытка («затопления») всех реагентов, кроме одного.
Катализ [ править ]

Изучение катализа и каталитических реакций очень важно для области физической органической химии. Катализатор участвует в химической реакции , но не расходуется в процессе. [12] [ нужна страница ] Катализатор снижает энергетический барьер активации (Δ G ‡ ), увеличивая скорость реакции либо за счет стабилизации структуры переходного состояния, либо за счет дестабилизации ключевого промежуточного продукта реакции, а поскольку требуется лишь небольшое количество катализатора, это может обеспечить экономичный доступ к дорогим или трудным для синтеза органическим молекулам. Катализаторы также могут влиять на скорость реакции, изменяя механизм реакции. [1] [ нужна страница ]
изотопный Кинетический эффект
Хотя закон скорости обеспечивает стехиометрию структуры переходного состояния , он не дает никакой информации о разрыве или образовании связей. [1] [ нужна страница ] Замещение изотопа вблизи реакционноспособного положения часто приводит к изменению скорости реакции. Изотопное замещение изменяет потенциальную энергию промежуточных продуктов реакции и переходных состояний, поскольку более тяжелые изотопы образуют более прочные связи с другими атомами. Атомная масса влияет на нулевое колебательное состояние связанных молекул, на более короткие и прочные связи в молекулах с более тяжелыми изотопами и на более длинные и слабые связи в молекулах с легкими изотопами. [6] [ нужна страница ] Поскольку колебательные движения часто меняются в ходе реакции из-за образования и разрыва связей, это влияет на частоты, и замена изотопа может дать представление о механизме реакции и законе скорости.
Эффекты заместителя [ править ]
Изучение того, как заместители влияют на реакционную способность молекулы или скорость реакций, представляет значительный интерес для химиков. Заместители могут оказывать влияние как через стерические , так и через электронные взаимодействия, последние из которых включают резонансный и индуктивный эффекты . молекулы Также можно повлиять на поляризуемость . Большинство эффектов заместителей анализируются с помощью линейных соотношений свободной энергии (LFER). Наиболее распространенным из них является анализ графика Хэммета . [1] [ нужна страница ] В этом анализе сравнивается влияние различных заместителей на ионизацию бензойной кислоты с их влиянием на различные химические системы. Параметры графиков Хэммета — сигма (σ) и ро (ρ). Значение σ указывает на кислотность замещенной бензойной кислоты по отношению к незамещенной форме. Положительное значение σ указывает на то, что соединение более кислое, а отрицательное значение указывает на то, что замещенная версия менее кислая. Значение ρ является мерой чувствительности реакции к изменению заместителя, но измеряет только индуктивные эффекты. Поэтому были созданы две новые шкалы, оценивающие стабилизацию локализованного заряда посредством резонанса. Один из них — σ + , который касается заместителей, которые стабилизируют положительные заряды посредством резонанса, а другой - σ − то есть для групп, которые стабилизируют отрицательные заряды посредством резонанса. Анализ Хэммета можно использовать для выяснения возможных механизмов реакции. Например, если предсказать, что структура переходного состояния имеет накопление отрицательного заряда по сравнению со структурой основного состояния, то можно ожидать, что электронодонорные группы увеличат скорость реакции. [1] [ нужна страница ]
и другие шкалы LFER Были разработаны . Стерические и полярные эффекты анализируются с помощью параметров Тафта . Замена растворителя вместо реагента может дать представление об изменениях заряда во время реакции. График Грюнвальда -Винштейна дает количественное представление об этих эффектах. [1] [ нужна страница ] [13]
Эффекты растворителя
Растворители могут оказывать сильное влияние на растворимость , стабильность и скорость реакции . Изменение растворителя также может позволить химику влиять на термодинамический или кинетический контроль реакции. В разных растворителях реакции протекают с разной скоростью из-за изменения распределения зарядов при химическом превращении. Эффекты растворителя могут воздействовать на структуры основного и/или переходного состояний . [1] [ нужна страница ]
Пример влияния растворителя на органические реакции можно увидеть при сравнении реакций SN 1 и SN 2 . [14] [ нужны дальнейшие объяснения ] [ нужен пример ]
Растворитель также может оказывать существенное влияние на термодинамическое равновесие системы, например, как в случае таутомеризации кето-енола . В неполярных апротонных растворителях енольная форма предпочтительнее из-за образования внутримолекулярной водородной связи , тогда как в полярных апротонных растворителях, таких как метиленхлорид , енольная форма менее предпочтительна из-за взаимодействия между полярным растворителем и полярный дикетон . [ нужен пример ] В протонных растворителях равновесие лежит в сторону кето-формы, поскольку внутримолекулярная водородная связь конкурирует с водородными связями, исходящими из растворителя. [15] [ нужен неосновной источник ] [ нужен неосновной источник ] [16] [ нужен неосновной источник ] [ нужен неосновной источник ] [17] [ нужен неосновной источник ] [ нужен неосновной источник ]

Современный пример изучения влияния растворителя на химическое равновесие увидеть в исследовании эпимеризации хиральных циклопропилнитрильных можно реактивов Гриньяра . [18] [ нужен неосновной источник ] [ нужен неосновной источник ] В этом исследовании сообщается, что константа равновесия цис - транс - изомеризации реактива Гриньяра намного больше - предпочтение цис- формы усиливается - в ТГФ в качестве растворителя реакции по сравнению с диэтиловым эфиром . Однако более высокая скорость цис-транс-изомеризации в ТГФ приводит к потере стереохимической чистоты. Это тот случай, когда понимание влияния растворителя на стабильность молекулярной конфигурации реагента важно с точки зрения селективности, наблюдаемой при асимметричном синтезе .
Квантовая химия [ править ]
Многие аспекты взаимосвязи структура-реактивность в органической химии можно объяснить с помощью резонанса , выталкивания электронов, индукции , правила восьми электронов и sp -гибридизации , но это всего лишь полезные формализмы, которые не отражают физическую реальность. Из-за этих ограничений истинное понимание физической органической химии требует более строгого подхода, основанного на физике элементарных частиц . Квантовая химия обеспечивает строгую теоретическую основу, позволяющую предсказывать свойства молекул посредством расчета электронной структуры молекулы, и она стала легкодоступным инструментом среди физико-химиков-органиков в виде популярных пакетов программного обеспечения. [ нужна ссылка ] Сила квантовой химии основана на волновой модели атома , в которой ядро представляет собой очень маленькую положительно заряженную сферу, окруженную диффузным электронным облаком. Частицы определяются связанной с ними волновой функцией — уравнением, которое содержит всю информацию, связанную с этой частицей. [12] [ нужна страница ] Вся информация о системе содержится в волновой функции. Эта информация извлекается из волновой функции с помощью математических операторов.

Энергия, связанная с определенной волновой функцией , возможно, самая важная информация, содержащаяся в волновой функции, может быть извлечена путем решения уравнения Шредингера (выше Ψ — волновая функция, E — энергия, а Ĥ — оператор Гамильтона) [12] [ нужна страница ] соответствующий гамильтонов оператор в котором применяется . В различных формах уравнения Шредингера общий размер распределения вероятностей частиц увеличивается с уменьшением массы частицы. По этой причине ядра имеют незначительный размер по сравнению с гораздо более легкими электронами и в практических приложениях квантовой химии рассматриваются как точечные заряды.
Из-за сложных взаимодействий, возникающих в результате электрон-электронного отталкивания, алгебраические решения уравнения Шредингера возможны только для систем с одним электроном, таких как атом водорода H 2 + , H3 2+ , и т. д.; однако из этих простых моделей возникают все знакомые атомные (s,p,d,f) и связывающие (σ,π) орбитали. В системах с несколькими электронами общая многоэлектронная волновая функция описывает все их свойства одновременно. Такие волновые функции генерируются путем линейного сложения одноэлектронных волновых функций для создания первоначального предположения, которое неоднократно модифицируется до тех пор, пока связанная с ним энергия не будет минимизирована. Часто требуются тысячи предположений, пока не будет найдено удовлетворительное решение, поэтому такие расчеты выполняются мощными компьютерами. Важно отметить, что решения для атомов с несколькими электронами дают такие свойства, как диаметр и электроотрицательность , которые точно отражают экспериментальные данные и закономерности, обнаруженные в таблице Менделеева . Решения для молекул, таких как метан , дают точное представление об их электронной структуре , которое невозможно получить экспериментальными методами. [ нужна ссылка ] Вместо четырех дискретных σ-связей между углеродом и каждым атомом водорода теория предсказывает набор из четырех связывающих молекулярных орбиталей, которые делокализованы по всей молекуле. Точно так же истинная электронная структура 1,3-бутадиена демонстрирует делокализованные с π-связями, молекулярные орбитали проходящие через всю молекулу, а не две изолированные двойные связи, как предсказывает простая структура Льюиса . [ нужна ссылка ]
Полная электронная структура обеспечивает большую предсказательную силу для органических преобразований и динамики, особенно в случаях, касающихся ароматических молекул , расширенных π-систем , связей между ионами металлов и органическими молекулами , молекул, содержащих нестандартные гетероатомы, такие как селен и бор , а также конформационной динамики больших молекул, таких как как белки , в которых многочисленные приближения в химических формализмах делают невозможным предсказание структуры и реакционной способности. Примером того, как определение электронной структуры является полезным инструментом для физико-химика-органика, является катализируемая металлами деароматизация бензола . Трикарбонил хрома обладает высокой электрофильностью из-за переноса электронной плотности с заполненных d-орбиталей хрома на разрыхляющие орбитали CO и способен ковалентно связываться с поверхностью молекулы бензола через делокализованные молекулярные орбитали . CO Лиганды индуктивно оттягивают электронную плотность бензола через атом хрома и резко активируют бензол, превращая его в нуклеофильная атака. Нуклеофилы затем могут реагировать с образованием гексациклодиенов, которые можно использовать в дальнейших преобразованиях, таких как циклоприсоединение Дильса-Альдера . [19]

Квантовая химия также может дать представление о механизме органического превращения без сбора каких-либо экспериментальных данных. Поскольку волновые функции обеспечивают полную энергию данного молекулярного состояния, предполагаемую молекулярную геометрию можно оптимизировать, чтобы получить релаксированные молекулярные структуры, очень похожие на те, которые обнаруживаются экспериментальными методами. [20] [ нужна страница ] координаты реакции Затем можно смоделировать переходного состояния и решить структуры . Таким образом, решение полной энергетической поверхности для данной реакции возможно, и такие расчеты применялись для решения многих задач органической химии, где кинетические данные недоступны или их трудно получить. [1] [ нужна страница ]
Спектроскопия, спектрометрия и кристаллография [ править ]
Физическая органическая химия часто предполагает определение молекулярной структуры, динамики и концентрации реагентов в ходе реакции. Взаимодействие молекул со светом может предоставить множество данных о таких свойствах посредством неразрушающих спектроскопических экспериментов , при этом свет поглощается, когда энергия фотона соответствует разнице в энергии между двумя состояниями в молекуле, и излучается, когда возбужденное состояние в молекуле коллапсирует. в более низкоэнергетическое состояние. Спектроскопические методы широко классифицируются по типу исследуемого возбуждения, например , колебательная , вращательная , электронная , спектроскопия ядерного магнитного резонанса (ЯМР) и спектроскопия электронного парамагнитного резонанса . Помимо спектроскопических данных, определению структуры часто помогают дополнительные данные, полученные в результате рентгеновской дифракции и масс-спектрометрических экспериментов. [21] [ нужна страница ]
ЯМР и ЭПР - спектроскопия

Одним из наиболее мощных инструментов физической органической химии является ЯМР-спектроскопия . Внешнее магнитное поле, приложенное к парамагнитному ядру, генерирует два дискретных состояния с положительными и отрицательными значениями спина, расходящимися по энергии ; затем разницу в энергии можно исследовать, определив частоту света, необходимую для возбуждения изменения спинового состояния для данного магнитного поля. Ядра, которые не являются неразличимыми в данной молекуле, поглощают на разных частотах, и интегрированная площадь пика в спектре ЯМР пропорциональна количеству ядер, реагирующих на эту частоту. [22] Можно количественно определить относительную концентрацию различных органических молекул просто путем интегрирования пиков в спектре, а многие кинетические эксперименты можно легко и быстро выполнить, отслеживая ход реакции в одном образце ЯМР. Протонный ЯМР часто используется химиками-синтетиками-органиками, поскольку протоны, связанные с определенными функциональными группами, характерные энергии поглощения, но ЯМР-спектроскопию также можно выполнять на изотопах азота дают , углерода , фтора , фосфора , бора и множества других элементов . В дополнение к простым экспериментам по поглощению также можно определить скорость реакций обмена быстрыми атомами посредством измерений подавления обмена, межатомных расстояний с помощью экспериментов по многомерному ядерному эффекту Оверхаузера и спин-спинового взаимодействия через связь посредством гомоядерной корреляционной спектроскопии . [23] Помимо свойств спинового возбуждения ядер, можно также изучать свойства органических радикалов с помощью того же фундаментального метода . Неспаренные электроны также имеют суммарный спин , а внешнее магнитное поле позволяет извлекать аналогичную информацию с помощью спектроскопии электронного парамагнитного резонанса (ЭПР). [1] [ нужна страница ]
Колебательная спектроскопия [ править ]
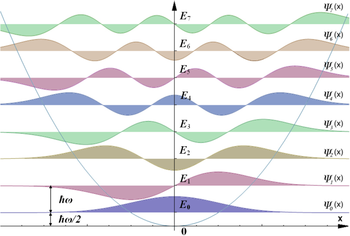
Колебательная спектроскопия , или инфракрасная (ИК) спектроскопия, позволяет идентифицировать функциональные группы и из-за своей низкой стоимости и надежности часто используется в учебных лабораториях и для мониторинга хода реакции в реальном времени в труднодоступных средах (высокое давление). , высокая температура, газовая фаза, фазовые границы ). Молекулярные колебания квантуются аналогично электронным волновым функциям, при этом целочисленное увеличение частоты приводит к переходам в более высокие энергетические состояния . Разница в энергии между колебательными состояниями почти постоянна, часто попадая в диапазон энергий, соответствующий инфракрасным фотонам, поскольку при нормальных температурах молекулярные колебания очень напоминают гармонические осцилляторы . Это позволяет грубо идентифицировать функциональные группы в органических молекулах , но спектры осложняются колебательной связью между соседними функциональными группами в сложных молекулах. Поэтому его полезность для определения структуры обычно ограничивается простыми молекулами. Еще больше усложняет ситуацию то, что некоторые вибрации не вызывают изменения в молекулярный дипольный момент и не будет наблюдаться с помощью стандартной ИК-спектроскопии поглощения. Вместо этого их можно исследовать с помощью рамановской спектроскопии , но этот метод требует более сложного оборудования и применяется реже. Однако, поскольку рамановская спектроскопия основана на рассеянии света, ее можно проводить на микроскопических образцах, таких как поверхность гетерогенного катализатора , граница фаз , или на подобразце объемом один микролитр (мкл) в большем объеме жидкости. [21] [ нужна страница ] Приложения колебательной спектроскопии часто используются астрономами для изучения состава облаков молекулярного газа , атмосфер внесолнечных планет и поверхностей планет .
Спектроскопия электронного возбуждения [ править ]
Спектроскопия электронного возбуждения , или ультрафиолетово-видимая (УФ-видимая) спектроскопия, выполняется в видимой и ультрафиолетовой областях электромагнитного спектра и полезна для исследования разницы в энергии между самой высокой занятой энергией (ВЗМО) и самой низкой незанятой энергией (НСМО). ) молекулярные орбитали . Эта информация полезна физикам-химикам-органикам при разработке органических фотохимических систем и красителей , поскольку поглощение видимого света различной длины придает цвет органическим молекулам . Таким образом, детальное понимание электронной структуры полезно для объяснения электронных возбуждений, а посредством тщательного контроля молекулярной структуры можно настроить зазор HOMO-LUMO для получения желаемых цветов и свойств возбужденного состояния. [24]
Масс-спектрометрия [ править ]
Масс-спектрометрия — это метод, который позволяет измерять молекулярную массу и предоставляет дополнительные данные к спектроскопическим методам структурной идентификации. В типичном эксперименте образец газовой фазы органического материала ионизируется , а полученные ионные частицы ускоряются приложенным электрическим полем в магнитное поле . Отклонение, создаваемое магнитным полем, часто в сочетании со временем, необходимым молекуле для достижения детектора, затем используется для расчета массы молекулы . Часто в ходе ионизации образца крупные молекулы распадаются, и в результирующих данных обнаруживается материнская масса и ряд более мелких фрагментных масс; такая фрагментация может дать глубокое понимание последовательности белков и полимеров нуклеиновых кислот. Помимо массы молекулы и ее фрагментов, также можно определить распределение масс изотопных вариантов и выявить качественное присутствие определенных элементов благодаря их характерному природному распределению изотопов. . Отношение массы фрагментов к численности родительских ионов можно сравнить с библиотекой эмпирических данных фрагментации и сопоставить с известной молекулярной структурой. [25] Комбинированная газовая хроматография и масс-спектрометрия используются для качественной идентификации молекул и количественного измерения концентрации с большой точностью и точностью, а также широко используются для проверки небольших количеств биомолекул и запрещенных наркотиков в образцах крови. Для химиков-синтетиков-органиков это полезный инструмент для характеристики новых соединений и продуктов реакций.
Кристаллография [ править ]
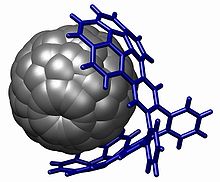
В отличие от спектроскопических методов, рентгеновская кристаллография всегда позволяет однозначно определить структуру и обеспечивает точные валентные углы и длины, которые совершенно недоступны при помощи спектроскопии. Он часто используется в физической органической химии для определения абсолютной молекулярной конфигурации и является важным инструментом для улучшения синтеза чистого энантиомерного вещества. Это также единственный способ определить положение и связь элементов, у которых отсутствует ЯМР, активное ядро например кислород . Действительно, до того, как в начале 20-го века стали доступны рентгеновские методы определения структуры, все органические структуры были полностью предположительными: тетраэдрический углерод , был подтвержден только кристаллической структурой алмаза например . [26] бензола была подтверждена кристаллической структурой гексаметилбензола . а делокализованная структура [27] Хотя кристаллография предоставляет химикам-органикам весьма удовлетворительные данные, она не является повседневным методом в органической химии, поскольку необходимо вырастить идеальный монокристалл целевого соединения. В этом методе нуждаются только сложные молекулы, для которых данные ЯМР не поддаются однозначной интерпретации. В приведенном ниже примере структуру комплекса хозяин-гость было бы довольно сложно решить без монокристаллической структуры: на фуллерене нет протонов и нет ковалентных связей между двумя половинками органического комплекса. не удалось доказать предполагаемую структуру. [ нужна ссылка ]
См. также [ править ]
- Журнал физической органической химии
- Гауссиан — пример используемого коммерчески доступного пакета программного обеспечения для квантовой механики. особенно в академических условиях
Ссылки [ править ]
- ^ Jump up to: Перейти обратно: а б с д и ж г час я дж к л м н тот п Догерти, Деннис А.; Анслин, Эрик В. (2006). Современная физико-органическая химия . Саусалито, Калифорния, США: Университетские научные книги. ISBN 9781891389313 . [ нужна страница ]
- ^ Тафт, РВ; Дено, Северная Каролина; Скелл, PS (октябрь 1958 г.). «Физическая органическая химия» . Ежегодный обзор физической химии . 9 (1): 287–314. Бибкод : 1958ARPC....9..287T . дои : 10.1146/annurev.pc.09.100158.001443 . ISSN 0066-426X .
- ^ Коэн, Н.; Бенсон, Юго-Запад (1 ноября 1993 г.). «Оценка теплот образования органических соединений аддитивными методами». Химические обзоры . 93 (7): 2419–2438. дои : 10.1021/cr00023a005 .
- ^ Бенсон, Сидни В.; Круикшанк, Франция; Золотой, ДМ; Хауген, Гилберт Р.; О'Нил, HE; Роджерс, А.С.; Шоу, Роберт; Уолш, Р. (1 июня 1969 г.). «Правила аддитивности для оценки термохимических свойств». Химические обзоры . 69 (3): 279–324. дои : 10.1021/cr60259a002 .
- ^ Кэри, Фрэнсис А. (2008). Органическая химия (7-е изд.). Бостон, Массачусетс, США: МакГроу-Хилл. ISBN 9780073047874 . [ нужна страница ]
- ^ Jump up to: Перейти обратно: а б Айзекс, Нил С. (1995). Физическая органическая химия (2-е изд.). Харлоу, ESS, ENG: Longman Scientific & Technical. ISBN 978-0582218635 . [ нужна страница ]
- ^ Мо, Йиронг; Гао, Цзяли (1 февраля 2007 г.). «Теоретический анализ вращательного барьера этана». Отчеты о химических исследованиях . 40 (2): 113–119. дои : 10.1021/ar068073w . ПМИД 17309192 . S2CID 16332261 .
- ^ Лю, Шубин (7 февраля 2013 г.). «Происхождение и природа барьеров вращения связей: единый взгляд». Журнал физической химии А. 117 (5): 962–965. Бибкод : 2013JPCA..117..962L . дои : 10.1021/jp312521z . ПМИД 23327680 .
- ^ Лю, Шубин; Говинд, Ниранджан (1 июля 2008 г.). «К пониманию природы барьеров внутреннего вращения с помощью новой схемы разделения энергии: этан и бутан». Журнал физической химии А. 112 (29): 6690–6699. Бибкод : 2008JPCA..112.6690L . дои : 10.1021/jp800376a . ПМИД 18563887 .
- ^ Ямамото, Такухей; Чен, Пи-Ю; Линь, Гуансинь; Блох-Мехкур, Анна; Якобсен, Нил Э.; Балли, Томас; Гласс, Ричард С. (1 октября 2012 г.). «Барьеры синтеза и вращения в 2,6-ди-(-анизил)анизоле» (PDF) . Журнал физической органической химии . 25 (10): 878–882. дои : 10.1002/poc.2939 .
- ^ Чен, Чен; Кэ, Цзиюань; Чжоу, X. Эдвард; Йи, Вэй; Брунзель, Джозеф С.; Ли, Цзюнь; Ён, Ю-Леонг; Сюй, Х. Эрик; Мельчер, Карстен (14 июля 2013 г.). «Структурные основы молекулярного распознавания фолиевой кислоты фолатными рецепторами» . Природа . 500 (7463): 486–489. Бибкод : 2013Natur.500..486C . дои : 10.1038/nature12327 . ПМК 5797940 . ПМИД 23851396 .
- ^ Jump up to: Перейти обратно: а б с д МакКуорри, Дональд А.; Саймон, Джон Д. (1997). Физическая химия: молекулярный подход (ред. Ред.). Саусалито, Калифорния, США: Университетские научные книги. ISBN 9780935702996 . Проверено 21 июня 2015 г. Обратите внимание, что доступ к этому тексту предоставляет Amazon, а не Google. [ нужна страница ]
- ^ Кевин, Деннис Н.; Д'Суза, Малкольм Дж. (1 июня 1992 г.). «О разработке весов ионизирующей способности растворителей на основе сольволиза бензильных субстратов». Журнал физической органической химии . 5 (6): 287–294. дои : 10.1002/poc.610050602 .
- ^ Райхардт, Кристиан; Велтон, Томас (2011). Растворители и эффекты растворителей в органической химии (4-е, обновленное и дополненное изд.). Вайнхайм, Германия: Wiley-VCH. ISBN 978-3-527-32473-6 .
- ^ Миллс, Сандер Г.; Бик, Питер (1 апреля 1985 г.). «Влияние растворителя на кето-енольное равновесие: испытания количественных моделей». Журнал органической химии . 50 (8): 1216–1224. дои : 10.1021/jo00208a014 .
- ^ Эмсли, Джон; Фриман, Невилл Дж. (1 октября 1987 г.). «Взаимодействия β-дикетонов». Журнал молекулярной структуры . 161 (1–2): 193–204. Бибкод : 1987JMoSt.161..193E . дои : 10.1016/0022-2860(87)85074-3 .
- ^ Шлунд, Себастьян; Базилио Янке, Элин М.; Вайс, Клаус; Энгельс, Бернд (1 января 2009 г.). «Прогнозирование таутомерного равновесия ацетилацетона в растворе. I. Правильный ответ по неправильной причине?». Журнал вычислительной химии . 31 (4): 665–70. дои : 10.1002/jcc.21354 . ПМИД 19557765 . S2CID 6003410 .
- ^ Jump up to: Перейти обратно: а б Гао, Мин; Патвардхан, Нирадж Н.; Карлье, Пол Р. (2013). «Стереохимическая инверсия цианостабилизированного реагента Гриньяра: замечательные эффекты структуры и концентрации эфирного растворителя». Дж. Ам. хим. Соц. 135 (38): 14390–14400. дои : 10.1021/ja407348s . ПМИД 23978216 .
- ^ Земмельхак, МФ; Холл, ХТ; Ёсифудзи, М. (сентябрь 1976 г.). «...Эта.5-Циклогексадиенилтрикарбонилхром(0) - интермедиаты в реакции карбанионов с -эта.6-арентрикарбонилхром(0)». Журнал Американского химического общества . 98 (20): 6387–6389. дои : 10.1021/ja00436a056 .
- ^ Шефер III, Генри Ф. (2004). Квантовая химия: развитие ab initio методов в теории молекулярной электронной структуры . Чикаго, Иллинойс, США: Р. Р. Доннелли (Курьер, Дувр). ISBN 978-0486432465 . Проверено 21 июня 2015 г. [ нужна страница ]
- ^ Jump up to: Перейти обратно: а б Драго, Рассел С. (1992). Физические методы для химиков (2-е изд.). футов. Уорт, Техас, США: Сондерс. ISBN 9780030970375 . Проверено 22 июня 2014 г. [ нужна страница ]
- ^ Джеймс Килер. «ЯМР и энергетические уровни (гл.2)» (PDF) . Понимание ЯМР-спектроскопии . Калифорнийский университет в Ирвайне . Проверено 26 октября 2013 г.
- ^ Киллер, Джеймс (2010). Понимание ЯМР-спектроскопии (2-е изд.). Чичестер: Уайли. ISBN 978-0-470-74608-0 .
- ^ Ройш, Уильям. «Видимая и ультрафиолетовая спектроскопия» . Веб-сайт Мичиганского государственного университета . Мичиганский государственный университет . Проверено 26 октября 2013 г.
- ^ Алан Дж. Хэндли; Эдвард Р. Адлард, ред. (2000). Газохроматографические методы и их применение . Бока-Ратон, Флорида: CRC Press. п. 168. ИСБН 978-0-8493-0514-6 .
- ^ Брэгг, Вашингтон; Брэгг, WL (июль 1913 г.). «Структура алмаза» . Природа . 91 (2283): 557. Бибкод : 1913Natur..91..557B . дои : 10.1038/091557a0 . S2CID 3987932 .
- ^ Лонсдейл, К. (ноябрь 1928 г.). «Строение бензольного кольца» . Природа . 122 (3082): 810. Бибкод : 1928Natur.122..810L . дои : 10.1038/122810c0 . S2CID 4105837 .
Дальнейшее чтение [ править ]
Общие [ править ]
- Питер Аткинс и Хулио де Паула, 2006, «Физическая химия», 8-е изд., Нью-Йорк, штат Нью-Йорк, США: Macmillan, ISBN 0716787598 , по состоянию на 21 июня 2015 г. [Например, см. стр. 422 для теоретико-группового/симметрического описания атомных орбиталей, способствующих связыванию в метане, CH 4 , и стр. 390f для оценки энергии связи π-электрона для 1,3-бутадиена по методу Хюккеля.]
- Томас Х. Лоури и Кэтлин Шуллер Ричардсон, 1987, Механизм и теория в органической химии, 3-е изд., Нью-Йорк, Нью-Йорк, США: Harper & Row, ISBN 0060440848 , по состоянию на 20 июня 2015 г. [Авторитетный учебник по этому предмету, содержащий ряд приложений, в которых представлены технические подробности теории молекулярных орбиталей, кинетических изотопных эффектов, теории переходного состояния и радикальной химии.]
- Эрик В. Анслин и Деннис А. Догерти, 2006, Современная физическая органическая химия , Саусалито, Калифорния: Университетские научные книги, ISBN 1891389319 . [Модернизированное и оптимизированное лечение с упором на приложения и междисциплинарные связи.]
- Майкл Б. Смит и Джерри Марч, 2007 г., «Продвинутая органическая химия Марта: реакции, механизмы и структура», 6-е изд., Нью-Йорк, штат Нью-Йорк, США: Wiley & Sons, ISBN 0470084944 , по состоянию на 19 июня 2015 г.
- Фрэнсис А. Кэри и Ричард Дж. Сундберг, 2006, «Продвинутая органическая химия: Часть A: Структура и механизмы», 4-е изд., Нью-Йорк, Нью-Йорк, США: Springer Science & Business Media, ISBN 0306468565 , по состоянию на 19 июня 2015 г.
- Хэммет, Луи П. (1940) Физическая органическая химия , Нью-Йорк, штат Нью-Йорк, США: McGraw Hill, по состоянию на 20 июня 2015 г.
История [ править ]
- Хаммонд, Джордж С. (1997). «Физическая органическая химия спустя 50 лет: она изменилась, но осталась ли она?» (PDF) . Чистое приложение. хим. 69 (9): 1919–22. дои : 10.1351/pac199769091919 . S2CID 53723796 . Проверено 20 июня 2015 г. [Выдающаяся отправная точка в истории этой области от чрезвычайно важного автора, ссылающегося и обсуждающего ранний текст Хэммета и т. д.]
Термохимия [ править ]
- Л.К. Дорайсвами , 2005, «Оценка свойств органических соединений (гл. 3),» стр. 36–51, 118–124 (ссылки), в журнале Organic Synthesis Engineering, Oxford, Oxon, ENG: Oxford University Press, ISBN 0198025696 , по состоянию на 22 июня 2015 г. (В этой главе книги рассматривается очень широкий спектр физических свойств и их оценка, включая узкий список термохимических свойств, представленный в статье WP за июнь 2015 г., в которой метод Бенсона и др. помещается рядом со многими другими методами. Л.К. Дорайсвами — заслуженный профессор инженерных наук Энсона Марстона в Университете штата Айова .)
- Ирикура, Карл К.; Фрурип, Дэвид Дж. (1998). «Вычислительная термохимия». В Ирикуре, Карл К.; Фрурип, Дэвид Дж. (ред.). Вычислительная термохимия: прогнозирование и оценка молекулярной термодинамики . Серия симпозиумов ACS. Том. 677. Американское химическое общество. стр. 2–18. дои : 10.1021/bk-1998-0677.ch001 . ISBN 978-0-8412-3533-5 .
Отрасли химии |
|---|