Карбанион
В органической химии карбанион анион — это , в котором углерод заряжен отрицательно. [1] [ не удалось пройти проверку ]
Формально карбанион — это сопряженное основание кислоты угольной :
- Р 3 СН + Б − → Р 3 С − + полупансион
где B обозначает основание . Карбанионы образуются в результате алканов ( sp при депротонирования 3 углерод), алкены (при пр. 2 углерод), арены (при пр. 2 углерод) и алкины (по атому углерода) известны как алкил- , алкенил ( винил ), арил и алкинил ( ацетилид ) анионы соответственно.
Карбанионы имеют концентрацию электронной плотности на отрицательно заряженном углероде, который в большинстве случаев эффективно реагирует с различными электрофилами различной силы, включая карбонильные группы , имины / соли иминия , галогенирующие реагенты (например, N -бромсукцинимид и дийод ). и доноры протонов . Карбанион — один из нескольких реакционноспособных промежуточных продуктов органической химии . В органическом синтезе литийорганические реагенты и реактивы Гриньяра обычно обрабатывают и называют «карбанионами». Это удобное приближение, хотя эти частицы обычно представляют собой кластеры или комплексы, содержащие высокополярные, но все же ковалентные связи металл-углерод (M д+ –С д- ), а не настоящие карбанионы.
Геометрия
[ редактировать ]В отсутствие π-делокализации отрицательный заряд карбаниона локализован в sp х гибридизованная орбиталь углерода в виде неподеленной пары . Как следствие, локализованные алкильные, алкенил/ариловые и алкинильные карбанионы принимают тригональную пирамидальную, изогнутую и линейную геометрию соответственно. Согласно правилу Бента , размещение карбанионной неподеленной пары электронов на орбитали со значительным s-характером является благоприятным, что объясняет пирамидальную и изогнутую геометрию алкильных и алкенильных карбанионов соответственно. Теория отталкивания электронных пар валентной оболочки (VSEPR) делает аналогичные предсказания. Это контрастирует с карбокатионами, которые отдают предпочтение незанятым несвязывающим орбиталям чистого атомного p-характера, что приводит к плоской и линейной геометрии соответственно для алкильных и алкенильных карбокатионов.


Однако делокализованные карбанионы могут отклоняться от этой геометрии. Вместо того, чтобы находиться на гибридной орбитали, неподеленная карбанионная пара может занимать ар-орбиталь (или орбиталь с высоким p-характером). p-орбиталь имеет более подходящую форму и ориентацию для перекрытия с соседней π-системой, что приводит к более эффективной делокализации заряда. Как следствие, алкилкарбанионы с соседними сопряженными группами (например, аллильные анионы, еноляты, нитронаты и т. д.) обычно имеют плоскую, а не пирамидальную форму. Точно так же делокализованные алкенилкарбанионы иногда предпочитают линейную, а не изогнутую геометрию. Чаще всего изогнутая геометрия по-прежнему предпочтительна для замещенных алкенил-анионов, хотя линейная геометрия лишь немного менее стабильна, что приводит к легкому уравновешиванию между ( E ) и ( Z )-изомерами (изогнутого) аниона через линейное переходное состояние . [2] Например, расчеты показывают, что исходный винил-анион или этиленид, Н 2 С=СН − , имеет инверсионный барьер 27 ккал/моль (110 кДж/моль), тогда как алленил-анион или алленид, Н 2 С=С=СН − ↔ Н 2 С − −C≡CH ), отрицательный заряд которого стабилизируется за счет делокализации, имеет инверсионный барьер всего 4 ккал/моль (17 кДж/моль), что отражает стабилизацию линейного переходного состояния за счет лучшей π-делокализации. [3]
Тенденции и возникновение
[ редактировать ]Карбанионы обычно являются нуклеофильными и основными. Основность и нуклеофильность карбанионов определяются заместителями при углероде. К ним относятся
- индуктивный эффект . Электроотрицательные атомы, прилегающие к заряду, стабилизируют заряд;
- степень сопряжения аниона. Резонансные эффекты могут стабилизировать анион. Это особенно верно, когда анион стабилизируется за счет ароматичности .
Геометрия также влияет на орбитальную гибридизацию несущего заряд карбаниона. Чем больше s-характер несущего заряд атома, тем стабильнее анион.
Карбанионы, особенно производные слабых углеродных кислот, которые не получают достаточного эффекта от двух перечисленных выше стабилизирующих факторов, обычно в разной степени чувствительны к кислороду и воде. В то время как некоторые из них просто разлагаются и разлагаются в течение нескольких недель или месяцев под воздействием воздуха, другие могут почти сразу же энергично и экзотермически реагировать с воздухом, приводя к самопроизвольному воспламенению ( пирофорность ). Среди карбанионных реагентов, часто встречающихся в лаборатории, ионные соли цианида водорода ( цианиды ) необычны тем, что они неопределенно стабильны в сухом воздухе и очень медленно гидролизуются в присутствии влаги.
Металлоорганические реагенты, такие как бутиллитий (гексамерный кластер, [BuLi] 6 ) или метилмагнийбромид (эфирный комплекс, MeMg(Br)(OEt 2 ) 2 ) часто называют «карбанионами», по крайней мере, в ретросинтетическом смысле. Однако на самом деле они представляют собой кластеры или комплексы, содержащие полярную ковалентную связь, хотя электронная плотность сильно поляризована в сторону атома углерода. Чем более электроположителен присоединенный атом металла, тем ближе поведение реагента к поведению настоящего карбаниона.
Фактически, настоящие карбанионы (т.е. виды, не присоединенные к стабилизирующему ковалентно связанному металлу) без электроноакцепторных и/или сопрягающих заместителей недоступны в конденсированной фазе, и эти виды необходимо изучать в газовой фазе. Некоторое время было неизвестно, могут ли простые алкил-анионы существовать в свободном виде; многие теоретические исследования предсказывали, что даже метанида анион CH - 3 должен быть несвязанным (т.е. сродство к электрону • CH 3 прогнозировался как отрицательный). Такая разновидность разложилась бы немедленно в результате спонтанного выброса электрона и, следовательно, была бы слишком мимолетной, чтобы ее можно было наблюдать непосредственно с помощью масс-спектрометрии. [4] Однако в 1978 г. метанид-анион был однозначно синтезирован путем воздействия на кетен электрического разряда, и сродство к электрону (EA) • CH 3 был определен с помощью фотоэлектронной спектроскопии как +1,8 ккал/моль, что делает его связанной разновидностью, но это лишь незначительно. Структура CH - 3 Было обнаружено, что имеет пирамидальную форму (C 3v ) с углом H-C-H 108 ° и инверсионным барьером 1,3 ккал / моль, в то время как • CH 3 был определен как плоский (точечная группа D 3h ). [5]
Простые первичные, вторичные и третичные зр. 3 карбанионы (например, этанид CH 3 CH - 2 , изопропанид (СН 3 ) 2 СН − и т -бутанид (СН 3 ) 3 С − впоследствии были определены как несвязанные виды (ЭА CH 3 CH 2 • , (СН 3 ) 2 СН• , (CH 3 ) 3 C• составляют -6, -7,4, -3,6 ккал/моль соответственно), что указывает на то, что α-замещение является дестабилизирующим. Однако относительно скромные стабилизирующие эффекты могут сделать их связанными. Например, циклопропильный и кубильный анионы связываются за счет увеличения s-характера орбитали неподеленной пары, тогда как неопентил- и фенетил -анионы также связываются в результате отрицательного гиперконъюгирования неподеленной пары с β-заместителем (n C → σ* С-С ). То же самое справедливо и для анионов с бензильной и аллильной стабилизацией. Газофазные карбанионы, представляющие собой sp 2 и sp-гибридизированные соединения гораздо более сильно стабилизированы и часто получаются непосредственно путем депротонирования в газовой фазе. [6]
В конденсированной фазе в качестве истинно ионных частиц были выделены только карбанионы, которые достаточно стабилизированы путем делокализации. В 1984 году Олмстед и Пауэр представили литий- краун-эфирную соль карбаниона трифенилметанида из трифенилметана , н -бутиллития и 12-краун-4 (который образует стабильный комплекс с катионами лития) при низких температурах: [7]

Formation of the triphenylmethane anion

Присоединение н -бутиллития к трифенилметану ( pKa ДМСО в CHPh 3 = 30,6) в ТГФ при низких температурах, а затем 12-краун-4 приводит к образованию красного раствора и солевого комплекса [Li(12-краун-4)] + [КФ 3 ] − выпадает в осадок при -20 ° C. Длина центральной связи C–C составляет 145 пм, фенильное кольцо движется под средним углом 31,2 °. Эта форма пропеллера менее выражена с противоионом тетраметиламмония. Кристаллическая структура аналогичного дифенилметанид-аниона ([Li(12-краун-4)] + [CHPh 2 ] − ), полученный из дифенилметана (p K a в ДМСО CH 2 Ph 2 = 32,3). Однако попытка выделения комплекса бензильного аниона PhCH - 2 из толуола (p K a в ДМСО CH 3 Ph ≈ 43) не увенчалась успехом из-за быстрой реакции образовавшегося аниона с растворителем ТГФ. [8] Свободный бензил-анион также был получен в фазе раствора импульсным радиолизом дибензилртути. [9]
В начале 1904 года [10] и 1917 год, [11] Шленк приготовил две соли красного цвета, сформулированные как [НМе 4 ] + [КФ 3 ] − и [НМе 4 ] + [ФЧ 2 ] − соответственно, метатезисом соответствующего натрийорганического реагента с хлоридом тетраметиламмония. Поскольку катионы тетраметиламмония не могут образовывать химическую связь с карбанионным центром, считается, что эти виды содержат свободные карбанионы. Хотя структура первого была подтверждена рентгеновской кристаллографией почти столетие спустя, [12] нестабильность последнего до сих пор препятствовала структурной проверке. Реакция предполагаемого» [НМе 4 ] + [ФЧ 2 ] − Сообщалось, что при взаимодействии с водой выделяется толуол и гидроксид тетраметиламмония, что является косвенным доказательством заявленного состава.
Одним из инструментов обнаружения карбанионов в растворе является протонный ЯМР . [13] Спектр циклопентадиена в ДМСО показывает четыре виниловых протона при 6,5 м.д. и два протона метиленового мостика при 3 м.д., тогда как циклопентадиенильный анион имеет одиночный резонанс при 5,50 м.д. Использование 6 Ли и 7 ЯМР лития предоставил данные о структуре и реакционной способности для различных видов литийорганических соединений .
Углеродные кислоты
[ редактировать ]Любое соединение, содержащее водород, в принципе может подвергаться депротонированию с образованием сопряженного основания. Соединение является угольной кислотой , если депротонирование приводит к потере протона у атома углерода. По сравнению с соединениями, которые обычно считаются кислотами (например, минеральными кислотами , такими как азотная кислота , или карбоновыми кислотами, такими как уксусная кислота ), угольные кислоты обычно на много порядков слабее, хотя существуют исключения (см. ниже). Например, бензол не является кислотой в классическом понимании Аррениуса , поскольку его водные растворы нейтральны. Тем не менее, это очень слабая кислота Бренстеда с предполагаемым p K a 49, которая может подвергаться депротонированию в присутствии супероснования, такого как основание Лохмана-Шлоссера ( н -бутиллитий и калия т- бутоксид ). Факторы, определяющие относительную устойчивость карбанионов, как сопряженных кислотно-основных пар, определяют также и упорядоченность значений p K a соответствующих карбоновых кислот. Кроме того, значения p K a позволяют предсказать, будет ли процесс переноса протона термодинамически выгодным: Б − чтобы быть термодинамически выгодным ( K соотношение p K a (BH) > p K a > 1), должно выполняться (AH).
Приведенные ниже значения представляют собой значения p K a , определенные в диметилсульфоксиде (ДМСО), который имеет более широкий полезный диапазон (от ~0 до ~35), чем значения, определенные в воде (от ~0 до ~14), и лучше отражает основность карбанионов в типичных органические растворители. Значения ниже 0 или больше 35 оцениваются косвенно; следовательно, численная точность этих значений ограничена. Значения p K a для водной среды также часто встречаются в литературе, особенно в контексте биохимии и энзимологии. Более того, водные значения часто приводятся во вводных учебниках по органической химии по педагогическим причинам, хотя вопрос зависимости от растворителей часто замалчивается. [14] В общем, значения p K a в воде и органическом растворителе значительно различаются, когда анион способен образовывать водородные связи. Например, в случае воды значения резко различаются: p K a в воде воды составляет 14,0, [15] тогда как p K a в ДМСО воды составляет 31,4, [16] что отражает различную способность воды и ДМСО стабилизировать гидроксид- анион. С другой стороны, для циклопентадиена численные значения сопоставимы: pKa в воде равно 15, а pKa в . ДМСО равно 18 [16]
Кислотность углекислого газа по p K a в ДМСО . [17]
Эти значения могут существенно отличаться от значений p K a для водной среды .Имя Формула Структурная формула п Ка в ДМСО Циклогексан С 6 Ч 12 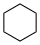
~60 Метан СН 4 
~56 Бензол С 6 Ч 6 
~49 [18] Пропен C3HC3H6 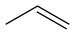
~44 Толуол С 6 Н 5 СН 3 
~43 Аммиак (N–H) НХ 3 
~41 Дитиан С 4 Ч 8 С 2 
~39 Диметилсульфоксид (СН 3 ) 2 ТАК 
35.1 Дифенилметан С 13 Ч 12 
32.3 Ацетонитрил СН 3 CN 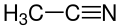
31.3 Анилин (N – H) С 6 Н 5 NH 2 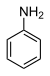
30.6 Трифенилметан С 19 Ч 16 
30.6 Фтороформ CHF3 швейцарских франка 
30.5 [19] Ксантен С 13 Н 10 О 
30.0 Этанол (O – H) С 2 Н 5 ОН 
29.8 Фенилацетилен С 8 Ч 6 
28.8 Тиоксантен С 13 Ч 10 С 
28.6 Ацетон С 3 Н 6 О 
26.5 Хлороформ CHClCHCl3 
24.4 [19] Бензоксазол С 7 Ч 5 НЕТ 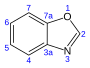
24.4 флуорен С 13 Ч 10 
22.6 Внутри C9HC9H8 
20.1 Циклопентадиен C5HC5H6 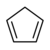
18.0 Нитрометан CH3NOCH3NO2 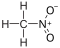
17.2 Диэтилмалонат С 7 Н 12 О 4 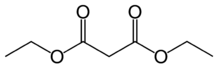
16.4 Ацетилацетон (H 3 CCO) 2 СН 2 
13.3 Цианистый водород HCN 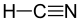
12.9 Уксусная кислота (O–H) СН 3 СООН 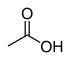
12.6 Малононитрилы C3H2NC3H2N2 
11.1 Димедон C8H12OC8H12O2 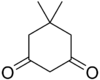
10.3 Кислота Мелдрама С 6 Н 8 О 4 
7.3 Гексафторацетилацетон (Ф 3 КСО) 2 СН 2 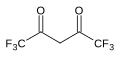
2.3 Хлороводород (Cl–H) HCl HCl (г) −2.0 [20] Трифлидовая кислота HC(SO 2 CF 3 ) 3 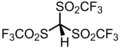
~ −16 [а]





















- Обратите внимание, что уксусная кислота, аммиак, анилин, этанол и хлористый водород не являются углекислотами, а являются обычными кислотами, показанными для сравнения.
- ^ Сообщаемое значение p K a в ацетонитриле (MeCN) составляет -3,7. [21] Величину p K a в ДМСО оценивали по корреляции p K a MeCN = 0,98 × р К а ДМСО + 11.6 . [22]
Как видно из приведенных выше примеров, кислотность увеличивается (p K a уменьшается), когда отрицательный заряд делокализован. Этот эффект возникает, когда заместители карбаниона ненасыщены и/или электроотрицательны. Хотя угольные кислоты обычно считаются кислотами, которые намного слабее, чем «классические» кислоты Бренстеда, такие как уксусная кислота или фенол, кумулятивный (аддитивный) эффект нескольких электроноакцепторных заместителей может привести к образованию кислот, которые будут такими же сильными или более сильными, чем неорганические минеральные кислоты. кислоты. Например, тринитрометан HC(NO 2 ) 3 , трицианометан HC(CN) 3 , пентацианоциклопентадиен C 5 (CN) 5 H и фульминовая кислота HCNO представляют собой сильные кислоты с водными значениями p K a , которые указывают на полный или почти полный перенос протона в воду. Трифлидовая кислота с тремя сильно электроноакцепторными трифлильными группами имеет расчетное значение p K a значительно ниже -10. На другом конце шкалы считается, что углеводороды, содержащие только алкильные группы, имеют значения p K a в диапазоне от 55 до 65. Таким образом, диапазон констант кислотной диссоциации углеродных кислот охватывает более 70 порядков величины.
Кислотность α-водорода в карбонильных соединениях позволяет этим соединениям участвовать в синтетически важных реакциях образования связей C–C, включая альдольную реакцию и присоединение Михаэля .
Хиральные карбанионы
[ редактировать ]С молекулярной геометрией карбаниона, описываемой как тригональная пирамида, вопрос заключается в том, могут ли карбанионы проявлять хиральность , потому что, если активационный барьер для инверсии этой геометрии слишком низок, любая попытка введения хиральности закончится рацемизацией , подобно азоту. инверсия . Однако существуют убедительные доказательства того, что карбанионы действительно могут быть хиральными, например, в исследованиях, проведенных с некоторыми литийорганическими соединениями.
Первые доказательства существования хиральных литийорганических соединений были получены в 1950 году. Реакция хирального 2-иодоктана с втор -бутиллитием в петролейном эфире при -70 ° C с последующей реакцией с сухим льдом дала в основном рацемическую 2-метилмасляную кислоту, но также и количество оптически активной 2-метилоктановой кислоты, которая могла образоваться только из столь же оптически активного 2-метилгептиллития с атомом углерода, связанным с карбанионом лития: [23]

Optically active organolithium

При нагревании реакции до 0°С оптическая активность теряется. Еще больше доказательств последовало в 1960-х годах. Реакция цис- изомера 2-метилциклопропилбромида с втор -бутиллитием снова с последующим карбоксилированием сухим льдом дала цис -2-метилциклопропилкарбоновую кислоту. Образование транс- изомера указывало бы на нестабильность промежуточного карбаниона. [24]

Stereochemistry of organolithiums

Таким же образом реакция (+)-( S ) -1 -бром- 1- метил-2,2-дифенилциклопропана с н- бутиллитием с последующей закалкой метанолом привела к получению продукта с сохранением конфигурации : [25]

Optical stability of 1-methyl-2,2-diphenylcyclopropyllithium

В последнее время появились хиральные соединения метиллития: [26]
![Хиральные окси[2H1]метиллитии. Bu означает бутил, i-Pr означает изопропил.](//upload.wikimedia.org/wikipedia/commons/thumb/1/1d/PhosphatePhosphonateRearrangement.png/500px-PhosphatePhosphonateRearrangement.png)
Chiral oxy[2H1]methyllithiums. Bu stands for butyl, i-Pr stands for isopropyl.
![Хиральные окси[2H1]метиллитии. Bu означает бутил, i-Pr означает изопропил.](http://upload.wikimedia.org/wikipedia/commons/thumb/1/1d/PhosphatePhosphonateRearrangement.png/500px-PhosphatePhosphonateRearrangement.png)
Фосфат содержит 1 хиральную группу с водородным и дейтериевым заместителями. Станнильная , группа замещается литием в интермедиат 2 , который подвергается фосфатно-фосфорановой перегруппировке в фосфоран 3 который при реакции с уксусной кислотой дает спирт 4 . И снова в диапазоне от -78 ° C до 0 ° C в этой реакционной последовательности сохраняется хиральность. ( Энантиоселективность определяли методом ЯМР-спектроскопии после дериватизации кислотой Мошера .)
История
[ редактировать ]Карбанионная структура впервые появилась в механизме реакции конденсации бензоина , правильно предложенном Кларком и Артуром Лэпвортом в 1907 году. [27] В 1904 году Вильгельм Шленк подготовил [Ф 3 С] − [НМе 4 ] + в поисках тетраметиламмония (из хлорида тетраметиламмония и Ph 3 CNa ) [10] а в 1914 году он продемонстрировал, как триарилметильные радикалы могут быть восстановлены до карбанионов щелочными металлами. [28] Фраза «карбанион» была введена Уоллисом и Адамсом в 1933 году как отрицательно заряженный аналог иона карбония. [29] [30]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Карбанион ». doi : 10.1351/goldbook.C00804
- ^ Карамелла, Пьерлуиджи; Хоук, КН (1 января 1981 г.). «Влияние электроноакцепторных заместителей на геометрию и барьеры инверсии виниловых анионов» . Буквы тетраэдра . 22 (9): 819–822. дои : 10.1016/0040-4039(81)80005-6 . ISSN 0040-4039 .
- ^ Алабугин, Игорь В. (19 сентября 2016 г.). Стереоэлектронные эффекты: мост между структурой и реактивностью . Чичестер, Великобритания: John Wiley & Sons, Ltd. doi : 10.1002/9781118906378 . ISBN 978-1-118-90637-8 .
- ^ Мэриник, Деннис С.; Диксон, Дэвид А. (1977). «Сродство к электрону метилового радикала: структуры CH 3 и CH −
3 " . Proceedings of the National Academy of Sciences of the United States of America . 74 (2): 410–413. Бибкод : 1977PNAS...74..410M . doi : 10.1073/pnas.74.2.410 . JSTOR 66197 . ПМЦ 392297 . ПМИД 16592384 . - ^ Эллисон, Дж. Барни; Энгелькинг, ПК; Линебергер, WC (апрель 1978 г.). «Экспериментальное определение геометрии и сродства к электрону метилового радикала». Журнал Американского химического общества . 100 (8): 2556–2558. дои : 10.1021/ja00476a054 . ISSN 0002-7863 .
- ^ Бланксби, С.Дж.; Боуи, Дж. Х. (2005). «Карбанионы: образование, строение и термохимия». Энциклопедия масс-спектрометрии . Гросс, Майкл Л., Каприоли, Р.М. (1-е изд.). Амстердам: Эльзевир. ISBN 9780080438504 . OCLC 55939535 .
- ^ Олмстед, Мэрилин М. (1985). «Выделение и рентгеноструктурные структуры литиевых краун-эфирных солей свободных фенилкарбанионов [CHPh 2 ] − и [CPh 3 ] − ". Журнал Американского химического общества . 107 (7): 2174–2175. doi : 10.1021/ja00293a059 .
- ^ Хардер, С. (2002). «Ранние «свободные» карбанионы Шленка». Химия: Европейский журнал . 8 (14): 3229–3232. doi : 10.1002/1521-3765(20020715)8:14<3229::AID-CHEM3229>3.0.CO;2-3 . ПМИД 12203352 .
- ^ Бократ, Брэдли; Дорфман, Леон М. (1 мая 2002 г.). «Субмикросекундное образование и наблюдение реакционноспособных карбанионов». Журнал Американского химического общества . 96 (18): 5708–5715. дои : 10.1021/ja00825a005 .
- ^ Перейти обратно: а б Шленк, В.; Вайкель, Т.; Герценштейн, А. (1910). «О трифенилметиле и аналогах трифенилметила в ряду бифенилов» . «Анналы химии» Юстуса Либиха . 372 : 1–20. дои : 10.1002/jlac.19103720102 .
- ^ Шленк, В.; Хольц, Джоанна (1917). «О бензилтетраметиламмонии» [О бензилтетраметиламмонии]. Отчеты Немецкого химического общества . 50 (1): 274–275. дои : 10.1002/cber.19170500143 . ISSN 1099-0682 .
- ^ Хардер, Сьерд (15 июля 2002 г.). «Ранние «свободные» карбанионы Шленка». Химия – Европейский журнал . 8 (14): 3229–3232. doi : 10.1002/1521-3765(20020715)8:14<3229::AID-CHEM3229>3.0.CO;2-3 . ПМИД 12203352 .
- ^ Касмаи, Хамид С. (июнь 1999 г.). «Простой и удобный метод генерации и ЯМР-наблюдения стабильных карбанионов». Журнал химического образования . 76 (6). дои : 10.1021/ed076p830 .
- ^ Хеллер, Стивен Т.; Сильверстайн, Тодд П. (23 апреля 2020 г.). «Значения pKa в учебной программе бакалавриата: введение значений pKa, измеренных в ДМСО, для иллюстрации эффектов растворителя» . Химтексты . 6 (2): 15. дои : 10.1007/s40828-020-00112-z . ISSN 2199-3793 .
- ^ Сильверстайн, Тодд П.; Хеллер, Стивен Т. (17 апреля 2017 г.). «Значения p K a в учебной программе бакалавриата: каково настоящее p K a воды?». Журнал химического образования . 94 (6): 690–695. Бибкод : 2017ЖЧЭд..94..690С . doi : 10.1021/acs.jchemed.6b00623 .
- ^ Перейти обратно: а б Эванс, Д.А.; Рипин, Д.Х. (2005). Chem 206 p K a « Таблица » (PDF) . Архивировано из оригинала (PDF) 2 июля 2019 г.
- ^ Бордвелл, Фредерик Г. (1988). «Равновесная кислотность в растворе диметилсульфоксида». Отчеты о химических исследованиях . 21 (12): 456–463. дои : 10.1021/ar00156a004 .
- ^ Бордвелл, Г.Ф.; Мэтьюз, Уолтер С. (1 мая 2002 г.). «Равновесные кислотности угольных кислот. III. Углекислоты в мембранном ряду». Журнал Американского химического общества . 96 (4): 1216–1217. дои : 10.1021/ja00811a041 .
- ^ Перейти обратно: а б Рассел, Джейми; Рокес, Николя (5 ноября 1998 г.). «Эффективное нуклеофильное трифторметилирование фтороформом и общим основанием». Тетраэдр . 54 (45): 13771–13782. дои : 10.1016/S0040-4020(98)00846-1 . ISSN 0040-4020 .
- ^ Трумаль, Александр; Липпинг, Лаури; Кальюранд, Ивари; Коппель, Ильмар А.; Лейто, Иво (06 мая 2016 г.). «Кислотность сильных кислот в воде и диметилсульфоксиде». Журнал физической химии А. 120 (20): 3663–3669. Бибкод : 2016JPCA..120.3663T . дои : 10.1021/acs.jpca.6b02253 . ПМИД 27115918 . S2CID 29697201 .
- ^ Хантер, Агнес; Родима, Томас; Мы можем, Яан; Книга, Элин; Горный лес, Вахур; Пляж Кальюр, Ивари; Коппель, Ильмар А.; Гарляускайте, Ромуте Ю.; Ягупольский Юрий Л.; Ягупольский Лев М.; Бернхардт, Эдуард; Виллнер, Хельге; Лейто, Иво (2011). «Равновесные кислотности суперкислот». Журнал органической химии . 76 (2): 391–395. дои : 10.1021/jo101409p . ПМИД 21166439 .
- ^ Дин, Фейчжи; Смит, Джереми М.; Ван, Хаобин (2009). «Основные принципы расчета значений p K a для органических кислот в неводном растворе». Журнал органической химии . 74 (7): 2679–2691. дои : 10.1021/jo802641r . ПМИД 19275192 .
- ^ Летсингер, Роберт Л. (1950). «Образование оптически активного 1-метилгептиллития». Журнал Американского химического общества . 72 (10): 4842. doi : 10.1021/ja01166a538 .
- ^ Эпплквист, Дуглас Э. (1961). «Конфигурационная стабильность цис- и транс -2-метилциклопропиллития и некоторые наблюдения по стереохимии их реакций с бромом и углекислым газом». Журнал Американского химического общества . 83 (4): 862–865. дои : 10.1021/ja01465a030 .
- ^ Валборский, HM (1964). «Циклопропаны. XV. Оптическая стабильность 1-метил-2,2-дифенилциклопропиллития». Журнал Американского химического общества . 86 (16): 3283–3288. дои : 10.1021/ja01070a017 .
- ^ Капеллер, Дагмар (2007). «Приготовление хирального α-окси-[ 2 H 1 ]метиллитий с 99% э.и. и определение их конфигурационной стабильности». Журнал Американского химического общества . 129 (4): 914–923. doi : 10.1021/ja066183s . PMID 17243828 .
- ^ Кларк, RWL; Лэпворт, А. (1907). «LXV. Расширение синтеза бензоина» . Журнал Химического общества, Сделки . 91 : 694–705. дои : 10.1039/CT9079100694 .
- ^ Шленк, В.; Маркус, Э. (1914). «О присоединении металлов к свободным органическим радикалам. XII. О триарилметиле» [О присоединении металлов к свободным органическим радикалам. XII. О триарилметиле». XII. триарилметилы]. Отчеты Немецкого химического общества . 47 (2): 1664. doi : 10.1002/cber.19140470256 .
- ^ Уоллис, ES; Адамс, Ф.Х. (1933). «Пространственная конфигурация валентностей в трехковалентных соединениях углерода1». Журнал Американского химического общества . 55 (9): 3838. doi : 10.1021/ja01336a068 .
- ^ Тидвелл, Т.Т. (1997). «Первый век физической органической химии: Пролог» . Чистая и прикладная химия . 69 (2): 211–214. дои : 10.1351/pac199769020211 . S2CID 98171271 .
Внешние ссылки
[ редактировать ]- Большая база данных значений Bordwell p K a на сайте www.chem.wisc.edu. Ссылка.
- Большая база данных значений Bordwell p K a на daecr1.harvard.edu. Ссылка
| Базы данных органов управления : Национальные |
|---|