Кинетический изотопный эффект
![{\displaystyle {\begin{matrix}\\{\ce {{CN^{-}}+{^{12}CH3-Br}->[k_{12}]{^{12}CH3-CN}+ Br^{-}}}\\{\ce {{CN^{-}}+{^{13}CH3-Br}->[k_{13}]{^{13}CH3-CN}+Br^ {-}}}\\{}\end{matrix}}\qquad {\text{KIE}}={\frac {k_{12}}{k_{13}}}=1,082\pm 0,008}](https://wikimedia.org/api/rest_v1/media/math/render/svg/438109ea220fd190ccc57f3e2c3726c47c24aae0)
В реакции бромистого метила с цианидом кинетический изотопный эффект углерода в метильной группе составил 1,082 ± 0,008. [1] [2]
В физической органической химии кинетический изотопный эффект ( КИЭ ) — изменение скорости химической реакции при одного из атомов реагирующих веществ замене одним из его изотопов . [3] Формально это отношение констант скорости реакций с участием легких ( k L ) и тяжелых ( k H ) изотопически замещенных реагентов (изотопологов):

Это изменение скорости реакции представляет собой квантово-механический эффект, который в первую очередь возникает из-за того, что более тяжелые изотопологи имеют более низкие частоты колебаний по сравнению с их более легкими аналогами. В большинстве случаев это подразумевает больший энергетический вклад, необходимый для достижения более тяжелыми изотопологами переходного состояния (или, в редких случаях, предела диссоциации ), и, следовательно, более медленную скорость реакции. Изучение кинетических эффектов изотопов может помочь в выяснении механизма некоторых химических реакций и иногда используется при разработке лекарств для улучшения неблагоприятной фармакокинетики путем защиты метаболически уязвимых связей CH.
Предыстория [ править ]
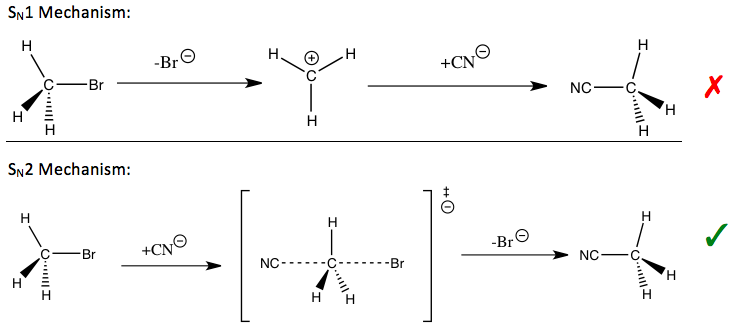
Кинетический изотопный эффект считается одним из наиболее важных и чувствительных инструментов изучения механизмов реакций , знание которого позволяет улучшить желаемые качества соответствующих реакций. Например, KIE можно использовать для определения того, нуклеофильного замещения протекает ли реакция по мономолекулярному (SN 1 ) или бимолекулярному (SN 2 ) пути.
В реакции бромистого метила и цианида (показана во введении) наблюдаемый метилуглерод КИЭ указывает на S N 2 -механизм. [1] В зависимости от пути можно использовать разные стратегии для стабилизации переходного состояния стадии , определяющей скорость реакции, а также повышения скорости и селективности реакции , что важно для промышленного применения.
Изменения изотопной скорости наиболее выражены, когда относительное изменение массы является наибольшим, поскольку эффект связан с частотами колебаний затронутых связей. Таким образом, заменяя обычный водород ( 1 H) с его изотопом дейтерием (D или 2 H) означает 100% увеличение массы; тогда как при замене углерода-12 на углерод-13 масса увеличивается всего на 8%. Скорость реакции с участием C– 1 Связь H обычно в 6–10 раз быстрее, чем связь C–. 2 H-связь, тогда как a 12 Реакция C протекает всего на 4% быстрее, чем соответствующая реакция. 13 реакция С; [4] : 445 хотя в обоих случаях изотоп на одну атомную единицу массы тяжелее.
Изотопное замещение может изменять скорость реакции различными способами. Во многих случаях разницу в скоростях можно объяснить, отметив, что масса атома влияет на частоту колебаний химической связи , которую он образует, даже если поверхность потенциальной энергии реакции почти идентична. Более тяжелые изотопы ( классически ) приведут к более низким частотам колебаний или, с точки зрения квантовой механики , будут иметь более низкую энергию нулевой точки . При более низкой энергии нулевой точки необходимо подать больше энергии для разрыва связи, что приводит к более высокой энергии активации разрыва связи, что, в свою очередь, снижает измеренную скорость (см., например, уравнение Аррениуса ). [3] [4] : 427
Классификация [ править ]
Первичные кинетические эффекты изотопные
Первичный кинетический изотопный эффект может быть обнаружен, когда образуется или разрывается связь с изотопно-меченым атомом. [3] [4] : 427 В зависимости от способа исследования KIE (параллельное измерение скоростей в сравнении с межмолекулярной конкуренцией и внутримолекулярной конкуренцией), наблюдение первичного KIE указывает на разрыв/образование связи с изотопом на этапе ограничения скорости или на последующий продукт. - определение шага(ов). (Заблуждение о том, что первичный KIE должен отражать разрыв/образование связи с изотопом на этапе ограничения скорости, часто повторяется в учебниках и основной литературе: см. раздел об экспериментах ниже. ) [5]
Для вышеупомянутых реакций нуклеофильного замещения первичные KIE были исследованы как для уходящих групп, нуклеофилов, так и для α-углерода, по которому происходит замещение. Интерпретация KIE уходящей группы поначалу была затруднена из-за значительного вклада факторов, не зависящих от температуры. КИЭ на α-углероде можно использовать для развития некоторого понимания симметрии переходного состояния в реакциях S N 2 , хотя этот КИЭ менее чувствителен, чем тот, который был бы идеальным, также из-за вклада невибрационных факторов. [1]
Вторичные кинетические эффекты изотопные
Вторичный кинетический изотопный эффект (или SKIE) наблюдается, когда связь с изотопно-меченным атомом в реагенте не разрывается и не образуется. [3] [4] : 427 Вторичные KIE, как правило, намного меньше первичных KIE; однако эффекты вторичного изотопа дейтерия могут достигать 1,4 на 2 Атом H, и были разработаны методы измерения эффектов изотопов тяжелых элементов с очень высокой точностью, поэтому вторичные KIE по-прежнему очень полезны для выяснения механизмов реакции.
Для вышеупомянутых реакций нуклеофильного замещения вторичные водородные KIE у α-углерода обеспечивают прямой способ отличить реакции SN 1 и SN 2 . Было обнаружено, что реакции S N 1 обычно приводят к большим вторичным KIE, приближающимся к своему теоретическому максимуму примерно 1,22, тогда как реакции S N 2 обычно приводят к вторичным KIE, которые очень близки или меньше 1. KIE, превышающие 1, называются нормальные кинетические изотопные эффекты , тогда как KIE меньше 1 называются обратными кинетическими изотопными эффектами . В общем, ожидается, что меньшие силовые константы в переходном состоянии дадут нормальный KIE, а большие силовые константы в переходном состоянии, как ожидается, дадут обратный KIE, когда вклады валентных колебаний доминируют над KIE. [1]
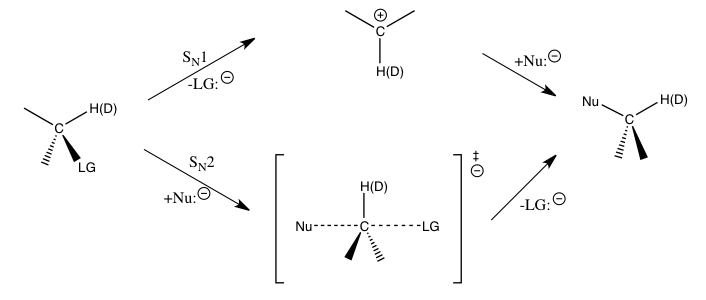
Величины таких вторичных изотопных эффектов на α-атоме углерода во многом определяются колебаниями Cα - H(D). Для реакции S N 1, поскольку атом углерода превращается в sp 2 гибридизированного иона карбения во время переходного состояния для определяющей скорость стадии с увеличением порядка связей Cα - H(D), можно было бы ожидать обратного КИЭ, если бы были важны только валентные колебания. Обнаружено, что наблюдаемые большие нормальные KIE вызваны значительными вкладами изгибных колебаний вне плоскости при переходе от реагентов к переходному состоянию образования ионов карбения. Для реакций S N 2 деформационные колебания по-прежнему играют важную роль для КИЭ, но вклады валентных колебаний имеют более сопоставимую величину, и результирующий КИЭ может быть нормальным или обратным в зависимости от конкретных вкладов соответствующих колебаний. [1] [6] [7]
Теория [ править ]
Теоретическая трактовка изотопных эффектов в значительной степени опирается на теорию переходного состояния , которая предполагает единую поверхность потенциальной энергии реакции и барьер между реагентами и продуктами на этой поверхности, поверх которого находится переходное состояние. [8] [9] KIE возникает в основном из-за изменений в основных колебательных состояниях, вызванных изотопическими возмущениями вдоль пути минимальной энергии поверхности потенциальной энергии, которые можно объяснить только с помощью квантово-механического подхода к системе. В зависимости от массы атома, движущегося по координате реакции, и природы (ширины и высоты) энергетического барьера, квантовое туннелирование также может вносить большой вклад в наблюдаемый кинетический изотопный эффект и, возможно, его придется рассматривать отдельно, помимо «полуклассическая» модель теории переходного состояния. [8]
Кинетический изотопный эффект дейтерия ( 2 H KIE) на сегодняшний день является наиболее распространенным, полезным и хорошо изученным типом KIE. Точный прогноз численного значения a 2 H KIE с использованием расчетов теории функционала плотности теперь является довольно рутинным. Более того, несколько качественных и полуколичественных моделей позволяют делать приблизительные оценки эффектов изотопа дейтерия без расчетов, часто предоставляя достаточно информации для рационализации экспериментальных данных или даже для подтверждения или опровержения различных механистических возможностей. Исходные материалы, содержащие дейтерий, часто коммерчески доступны, что делает синтез изотопно-обогащенных исходных материалов относительно простым. Кроме того, из-за большой относительной разницы в массе 2 Рука 1 H и сопутствующих различий в частоте колебаний, величина изотопного эффекта больше, чем у любой другой пары изотопов, за исключением 1 Рука 3 ЧАС, [10] позволяя легко измерять и интерпретировать как первичные, так и вторичные изотопные эффекты. Напротив, вторичные эффекты для более тяжелых элементов обычно очень малы и близки по величине к экспериментальной неопределенности, что усложняет их интерпретацию и ограничивает их полезность. В контексте изотопных эффектов водород часто используется для обозначения легкого изотопа протия ( 1 Х), конкретно. В оставшейся части статьи упоминание водорода и дейтерия в параллельных грамматических конструкциях или прямое сравнение между ними следует интерпретировать как обращение к протию и дейтерию. [а]
Теория KIE была впервые сформулирована Якобом Бигелайзеном в 1949 году. [11] [4] : 427 Общая формула Биглейзена для 2 H KIE (который также применим к более тяжелым элементам) приведен ниже. Он использует теорию переходного состояния и статистическую механическую обработку поступательных, вращательных и колебательных уровней для расчета констант скорости k H и k D . Однако эта формула является «полуклассической» в том смысле, что в ней не учитывается вклад квантового туннелирования, который часто вводится как отдельный поправочный коэффициент. Формула Биглейзена также не учитывает различия в несвязанных отталкивающих взаимодействиях, вызванные немного более короткой связью C–D по сравнению со связью C–H. В уравнении количества с индексами H или D относятся к замещенным водородом или дейтерием частицам соответственно, а количества с двойным крестиком ‡ или без него относятся к переходному состоянию или основному состоянию реагента соответственно. [7] [12] (Строго говоря, Следует также включить термин, возникающий из-за изотопной разницы в коэффициентах передачи. [13] )

- ,
![{\displaystyle {\frac {k_{{\ce {H}}}}{k_{{\ce {D}}}}}=\left({\frac {\sigma _{{\ce {H}} }\sigma _{{\ce {D}}}^{\ddagger }}{\sigma _{{\ce {D}}}\sigma _{{\ce {H}}}^{\ddagger }} }\right)\left({\frac {M_{{\ce {H}}}^{\ddagger }M_{{\ce {D}}}}{M_{{\ce {D}}}^{ \ddagger }M_{{\ce {H}}}}}\right)^{\frac {3}{2}}\left({\frac {I_{x{\ce {H}}}^{\ ddagger }I_{y{\ce {H}}}^{\ddagger }I_{z{\ce {H}}}^{\ddagger }}{I_{x{\ce {D}}}^{\ ddagger }I_{y{\ce {D}}}^{\ddagger }I_{z{\ce {D}}}^{\ddagger }}}{\frac {I_{x{\ce {D}} }I_{y{\ce {D}}}I_{z{\ce {D}}}}{I_{x{\ce {H}}}I_{y{\ce {H}}}I_{z {\ce {H}}}}}\right)^{\frac {1}{2}}\left({\frac {\prod \limits _{i=1}^{3N^{\ddagger }- 7}{\frac {1-e^{-u_{i{\ce {D}}}^{\ddagger }}}{1-e^{-u_{i{\ce {H}}}^{ \ddagger }}}}}{\prod \limits _{i=1}^{3N-6}{\frac {1-e^{-u_{i{\ce {D}}}}}{1- e^{-u_{i{\ce {H}}}}}}}}\right)e^{-{\frac {1}{2}}\left[\sum \limits _{i=1} ^{3N^{\ddagger }-7}(u_{i{\ce {H}}}^{\ddagger }-u_{i{\ce {D}}}^{\ddagger })-\sum \ пределы _{i=1}^{3N-6}(u_{i{\ce {H}}}-u_{i{\ce {D}}})\right]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/93f26faede9d0fba35d6f675e641c716e7284c0d)
где мы определяем
- и .


Здесь h — постоянная Планка , k B — постоянная Больцмана , — частота вибрации, выраженная в волновых числах , c — скорость света , NA — , постоянная Авогадро а R — универсальная газовая постоянная . σ X (X = H или D) — числа симметрии реагентов и переходных состояний. MX — члены молекулярные массы соответствующих видов, а I q X ( q = x , y или z ) — моменты инерции относительно трех главных осей. u вибрации (ZPE) (см . i X прямо пропорциональны соответствующим частотам колебаний ν i и энергии нулевой точки ниже). Целые числа N и N ‡ – число атомов в реагентах и переходных состояниях соответственно. [7] Сложное выражение, приведенное выше, можно представить как произведение четырех отдельных факторов: [7]

- .

В частном случае эффектов изотопа дейтерия мы будем утверждать, что первые три члена можно рассматривать как равные или хорошо аппроксимируемые единицей. Первый фактор S (содержащий σ X ) представляет собой соотношение чисел симметрии различных видов. Это будет рациональное число (отношение целых чисел), зависящее от количества вращений молекул и связей, приводящих к перестановке одинаковых атомов или групп в реагентах и переходному состоянию. [12] Для систем низкой симметрии все σ X (реагент и переходное состояние) будут равны единице; таким образом, S часто можно пренебречь. Коэффициент MMI (содержащий M X и I q X ) относится к соотношению молекулярных масс и моментов инерции. Поскольку водород и дейтерий, как правило, намного легче, чем большинство реагентов и переходных состояний, существует небольшая разница в молекулярных массах и моментах инерции между молекулами, содержащими H и D, поэтому коэффициент MMI обычно также приближается к единице. Фактор EXC (содержащий произведение колебательных статистических сумм ) корректирует KIE, вызванный реакциями колебательно-возбужденных молекул. Доля молекул, обладающих достаточной энергией, чтобы иметь колебания связи A–H/D в возбужденном состоянии, обычно невелика для реакций при комнатной температуре или около нее (связи с водородом обычно вибрируют при 1000 см-1). −1 или выше, поэтому exp(- u i ) = exp(- hν i / k B T ) < 0,01 при 298 К, что приводит к незначительному вкладу факторов 1 – exp(- u i ). Следовательно, для KIE водорода/дейтерия в наблюдаемых значениях обычно доминирует последний фактор, ZPE (экспоненциальная функция колебательных различий ZPE), состоящий из вкладов различий ZPE для каждой из колебательных мод реагентов и переходного состояния, что можно представить следующим образом: [7]
- ,
![{\displaystyle {\begin{aligned}{\frac {k_ {{\ce {H}}}}{k_ {{\ce {D}}}}}&\cong \exp \left\{- {\frac {1}{2}}\left[\sum \limits _{i=1}^{3N^{\ddagger }-7}(u_{i{\ce {H}}}^{\ddagger }-u_ {i{\ce {D}}}^{\ddagger })-\sum \limits _{i=1}^{3N-6}(u_{i{\ce {H}}}-u_{i{ \ce {D}}})\right]\right\}\\&\cong \exp \left[\sum _{i}^{\mathrm {(react.)} }{\frac {1}{2 }}\Delta u_{i}-\sum _{i}^{\mathrm {(TS)} }{\frac {1}{2}}\Delta u_{i}^{\ddagger }\right]\ конец {выровнено}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/20d669d624e10fcb89d116442c5543998ea4fbf8)
где мы определяем
- и .


Суммы в показателе степени второго выражения можно интерпретировать как пробегающие все колебательные моды основного состояния реагента и переходного состояния. Альтернативно их можно интерпретировать как прохождение тех мод, которые присущи только реагенту или переходному состоянию или частоты колебаний которых существенно изменяются при продвижении по координате реакции. Остальные пары колебательных мод реагента и переходного состояния имеют очень схожие характеристики. и , и отмены происходят при вычислении сумм в показателе степени. Таким образом, на практике KIE дейтерия часто в значительной степени зависят от нескольких ключевых колебательных мод из-за этого подавления, что делает качественный анализ k H / k D. возможным [12]


Как уже упоминалось, особенно при замещении водорода/дейтерия, большинство KIE возникают из-за разницы в ZPE между реагентами и переходным состоянием рассматриваемых изотопологов, и эту разницу можно понять качественно с помощью следующего описания: в рамках приближения Борна- Оппенгеймера поверхность потенциальной энергии одинакова для обоих изотопов. Однако квантовомеханическая трактовка энергии вводит на эту кривую дискретные колебательные уровни, и наименьшее возможное энергетическое состояние молекулы соответствует наименьшему колебательному энергетическому уровню, который немного выше по энергии, чем минимум кривой потенциальной энергии. Эта разница, известная как ZPE, является проявлением принципа неопределенности Гейзенберга, который требует неопределенности в длине связи CH или CD. Поскольку более тяжелый (в данном случае дейтерированный) вид ведет себя более «классически», его уровни колебательной энергии ближе к классической кривой потенциальной энергии и он имеет более низкий ZPE. Различия ZPE между двумя видами изотопов, по крайней мере, в большинстве случаев, уменьшаются в переходном состоянии, поскольку константа силы связи уменьшается во время разрыва связи. Следовательно, более низкий ZPE дейтерированных частиц приводит к большей энергии активации реакции, как показано на следующем рисунке, что приводит к нормальному KIE. [14] Этот эффект в принципе следует учитывать для всех 3 N− 6 мод колебаний исходного материала и 3 N ‡ − 7 мод колебаний в переходном состоянии (одна мода, соответствующая координате реакции, в переходном состоянии отсутствует, так как связь разрывается и нет восстанавливающей силы, препятствующей движению). Гармонический осциллятор является хорошим приближением для колеблющейся связи, по крайней мере, для колебательных состояний с низкой энергией. Квантовая механика дает колебательное ZPE как . Таким образом, мы можем легко интерпретировать фактор 1/2 и суммы термины по колебательным модам основного состояния и переходного состояния в показателе степени упрощенной формулы, приведенной выше. Для гармонического осциллятора частота колебаний обратно пропорциональна корню квадратному из приведенной массы колебательной системы:


- ,

где k f — силовая постоянная . Более того, приведенная масса аппроксимируется массой легкого атома системы X = H или D. Поскольку m D составляет примерно 2 m H ,
- .

В случае гомолитической диссоциации связи C–H/D член переходного состояния исчезает, и в пренебрежении другими колебательными модами k H / k D = exp( 1 / 2 Δ ты я ). Таким образом, больший изотопный эффект наблюдается для более жесткой («более прочной») связи C–H/D. В большинстве представляющих интерес реакций атом водорода переносится между двумя атомами с переходным состоянием [A···H···B] ‡ и необходимо учитывать колебательные моды в переходном состоянии. Тем не менее, в целом верно, что разрыв связи с более высокой частотой колебаний даст больший изотопный эффект.

Для расчета максимально возможного значения для нетуннельного дейтериевого КИЭ рассмотрим случай, когда разность ЗПЭ между валентными колебаниями типичной углерод-водородной связи (3000 см-1) −1 ) и связь углерод-дейтерий (2200 см −1 ) исчезает в переходном состоянии (разница энергий (1/2)(3000 – 2200 см −1 ) = 400 см −1 или около 1,15 ккал/моль), без какой-либо компенсации за счет нулевой разницы энергий в переходном состоянии (например, из-за симметричного растяжения A···H···B, которое уникально для переходного состояния). Упрощенная формула, приведенная выше, предсказывает максимум k H / k D как 6,9. Если учесть полное исчезновение двух изгибных колебаний, то можно прогнозировать значения k H / k D , достигающие 15-20. Однако маловероятно, что частоты изгиба исчезнут в переходном состоянии, и есть лишь несколько случаев, когда значения k H / k D превышают 7-8 вблизи комнатной температуры. Более того, часто обнаруживается, что туннелирование является основным фактором, когда они превышают эти значения. Значение k H / k D ~ 10 считается максимальным для полуклассического первичного кинетического изотопного эффекта (без туннелирования) для реакций, протекающих около 298 К. (Формула для k H / k D имеет температурную зависимость, поэтому большие изотопные эффекты возможны при более низких температурах). [15] В зависимости от характера переходного состояния H-переноса (симметричного или «раннего» или «позднего» и линейного или изогнутого) степень приближения эффекта первичного изотопа дейтерия к этому максимуму варьируется. Модель, разработанная Вестхаймером, предсказала, что симметричные (термонейтральные, по постулату Хаммонда ) линейные переходные состояния обладают наибольшими изотопными эффектами, тогда как переходные состояния, которые являются «ранними» или «поздними» (для экзотермических или эндотермических реакций соответственно), или нелинейными ( например, циклический) проявляют меньшие эффекты. Эти предсказания с тех пор получили обширную экспериментальную поддержку. [16]
Что касается эффектов вторичного изотопа дейтерия, Стрейтвизер предположил, что ослабление (или усиление, в случае обратного изотопного эффекта) изгибных мод от основного состояния реагента к переходному состоянию в значительной степени ответственно за наблюдаемые изотопные эффекты. Эти изменения объясняются изменением стерического окружения, когда углерод, связанный с H/D, подвергается регибридизации из sp. 3 sp 2 или наоборот (вторичный кинетический изотопный эффект α) или ослабление связи из-за гиперконъюгации в случаях, когда карбокатион образуется на расстоянии одного атома углерода (вторичный β-KIE). Эти изотопные эффекты имеют теоретический максимум k H / k D = 2. 0.5 ≈ 1,4. Для SKIE в положении α регибридизация от sp 3 sp 2 дает нормальный изотопный эффект, а регибридизация от sp 2 sp 3 приводит к обратному изотопному эффекту с теоретическим минимумом k H / k D = 2 -0.5 ≈ 0,7. На практике k H / k D ~ 1,1-1,2 и k H /k D ~ 0,8-0,9 характерны для α-вторичных кинетических изотопных эффектов, тогда как k H / k D ~ 1,15-1,3 характерны для β-вторичных кинетических изотопных эффектов. Для реагентов, содержащих несколько изотопно-замещенных β-водородов, наблюдаемый изотопный эффект часто является результатом согласованного действия нескольких H/D в β-положении. В этих случаях эффект каждого изотопно-меченого атома является мультипликативным, и случаи, когда k H / k D > 2, нередки. [17]
Следующие простые выражения, связывающие KIE дейтерия и трития, которые также известны как уравнение Суэйна (или уравнения Суэйна-Шаада-Стиверса), могут быть получены из общего выражения, приведенного выше, с использованием некоторых упрощений: [8] [18]
- ;

то есть,
- .

При выводе этих выражений разумное приближение заключается в том, что приведенная масса примерно равна массе тела. 1 ЧАС, 2 Н или 3 Х, использовался. При этом предполагалось, что колебательное движение аппроксимируется гармоническим осциллятором, так что (X = H, D или T). Индекс « s » относится к этим «полуклассическим» кинетическим изотопным эффектам, которые игнорируют квантовое туннелирование. Вклад туннелирования следует рассматривать отдельно как поправочный коэффициент.

Для изотопных эффектов, включающих элементы, отличные от водорода, многие из этих упрощений недействительны, и величина изотопного эффекта может сильно зависеть от некоторых или всех пренебрегаемых факторов. Таким образом, KIE для элементов, отличных от водорода, часто гораздо сложнее рационализировать или интерпретировать. Во многих случаях, особенно в реакциях переноса водорода, вклад туннелирования в KIE значителен (см. ниже).
Туннелирование [ править ]
В некоторых случаях наблюдается дополнительное увеличение скорости для более легкого изотопа, возможно, из-за квантового туннелирования . Обычно это наблюдается только для реакций, включающих связи с водородом. Туннелирование происходит, когда молекула проникает через потенциальный энергетический барьер, а не через него. [19] [20] Хотя это и не разрешено классической механикой , частицы могут проходить через классически запрещенные области пространства в квантовой механике, основанной на дуализме волна-частица . [21]
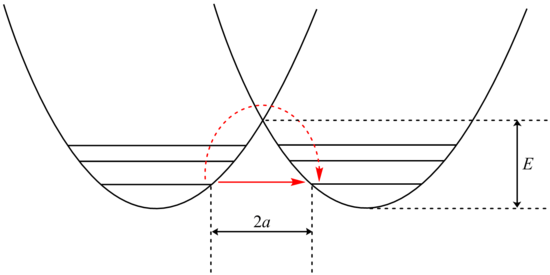
Туннелирование можно проанализировать с помощью модификации Белла уравнения Аррениуса , которая включает добавление туннельного коэффициента Q:

где A — параметр Аррениуса, E — высота барьера и

где и


Исследование члена β показывает экспоненциальную зависимость от массы частицы. В результате туннелирование гораздо более вероятно для более легких частиц, таких как водород. Простое удвоение массы туннелирующего протона путем замены его дейтроном резко снижает скорость таких реакций. В результате наблюдаются очень большие КИЭ, которые нельзя объяснить различиями в ЗПЭ.

Кроме того, член β линейно зависит от ширины барьера 2a. Как и в случае с массой, туннелирование наиболее эффективно при небольшой ширине барьера. Оптимальные расстояния туннелирования протонов между атомом донора и акцептора составляют 0,4 Å. [23]



изотопный кинетический Переходный эффект
Изотопный эффект, выраженный приведенными выше уравнениями, относится только к реакциям, которые можно описать кинетикой первого порядка . Во всех случаях, когда это невозможно, переходные KIE с использованием уравнений GEBIK и GEBIF. следует учитывать [34]
Эксперименты [ править ]
Симмонс и Хартвиг называют следующие три случая основными типами экспериментов по кинетическому изотопному эффекту, включающим функционализацию связи CH: [5]
- А) КИЭ определяется по абсолютным скоростям двух параллельных реакций

В этом эксперименте константы скорости для нормального субстрата и его меченого изотопами аналога определяются независимо, а KIE получается как отношение этих двух. Точность измерения KIE сильно ограничена точностью, с которой может быть измерена каждая из этих констант скорости. Более того, воспроизведение точных условий в двух параллельных реакциях может быть очень сложной задачей. Тем не менее, измерение большого кинетического изотопного эффекта путем прямого сравнения констант скорости указывает на то, что разрыв связи CH происходит на этапе, определяющем скорость. (Меньшее значение может указывать на изотопный эффект из-за предварительного равновесия, так что разрыв связи CH происходит где-то до этапа, определяющего скорость.)
- Б) КИЕ определяется в результате межмолекулярной конкуренции
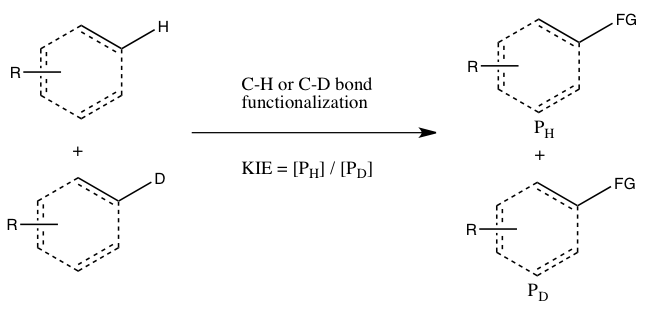
В экспериментах этого типа используются те же субстраты, которые использовались в эксперименте А, но им разрешается вступать в реакцию в одном и том же контейнере, а не в двух отдельных контейнерах. Кинетический изотопный эффект в этом эксперименте определяется относительным количеством продуктов, образовавшихся в результате функционализации CH и CD (или его можно вывести из относительных количеств непрореагировавших исходных материалов). Чтобы наблюдать кинетический изотопный эффект, необходимо погасить реакцию до ее завершения (см. раздел «Оценка» ниже). Обычно реакцию останавливают при низкой конверсии (конверсия от ~5 до 10%) или при использовании большого избытка (>5 экв.) изотопной смеси. Этот тип эксперимента гарантирует, что функционализация связей CH и CD происходит в одних и тех же условиях, а соотношение продуктов функционализации связей CH и CD можно измерить с гораздо большей точностью, чем константы скорости в эксперименте A. Более того, только одно измерение требуется концентрация продукта из одного образца. Однако наблюдаемый кинетический изотопный эффект в этом эксперименте труднее интерпретировать, поскольку он может означать, что разрыв связи CH происходит либо во время этапа определения скорости, либо на этапе определения продукта, следующем за этапом определения скорости. Отсутствие кинетического изотопного эффекта, по крайней мере, по мнению Симмонса и Хартвига, тем не менее указывает на то, что разрыв связи CH не происходит на стадии определения скорости.
- В) КИЭ определяется в результате внутримолекулярной конкуренции
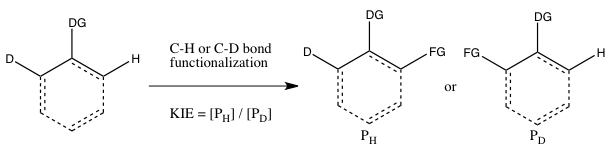
Этот тип эксперимента аналогичен эксперименту B, за исключением того, что на этот раз происходит внутримолекулярная конкуренция за функционализацию связей CH или CD. В большинстве случаев субстрат имеет направляющую группу (DG) между связями CH и CD. Расчет кинетического изотопного эффекта в этом эксперименте и его интерпретация основаны на тех же соображениях, что и в эксперименте B. Однако результаты экспериментов B и C будут отличаться, если необратимое связывание содержащего изотоп субстрата происходит в эксперименте до B разрыв связи CH или CD. В таком сценарии изотопный эффект может наблюдаться в эксперименте C (где выбор изотопа может происходить даже после связывания субстрата), но не в эксперименте B (поскольку выбор того, будет ли разрываться связь CH или CD, уже сделан, как только субстрат связывается необратимо). В отличие от эксперимента Б, реакцию не нужно останавливать при низком потреблении изотопного исходного материала для получения точного результата. k H / k D , поскольку соотношение H и D в исходном материале составляет 1:1 независимо от степени превращения.
Одним из примеров различных изотопных эффектов, не связанных с активацией CH, наблюдаемых в случае межмолекулярной (эксперимент B) и внутримолекулярной (эксперимент C) конкуренции, является фотолиз дифенилдиазометана в присутствии трет -бутиламина. Для объяснения этого результата было предложено образование дифенилкарбена с последующей необратимой нуклеофильной атакой трет -бутиламина. Поскольку существует небольшая изотопная разница в скорости нуклеофильной атаки, межмолекулярный эксперимент привел к KIE, близкому к 1. Однако во внутримолекулярном случае соотношение продуктов определяется переносом протона, который происходит после нуклеофильной атаки, процесс для где имеется существенный KIE 2,6. [35]

Таким образом, эксперименты A, B и C дадут результаты разного уровня точности и потребуют разных экспериментальных установок и способов анализа данных. В результате осуществимость каждого типа эксперимента будет зависеть от кинетического и стехиометрического профиля реакции, а также физических характеристик реакционной смеси (например, гомогенной или гетерогенной). Более того, как отмечалось в абзаце выше, эксперименты предоставляют данные о кинетическом изотопном эффекте для различных стадий многостадийной реакции, в зависимости от относительного расположения лимитирующей стадии, стадий, определяющих продукт, и/или расщепления CH/D. шаг.
Гипотетические примеры ниже иллюстрируют распространенные сценарии. Рассмотрим следующую диаграмму координат реакции. Для реакции с этим профилем все три эксперимента (A, B и C) дадут значительный первичный кинетический изотопный эффект:

С другой стороны, если реакция следует следующему энергетическому профилю, в котором разрыв связи CH или CD необратим, но происходит после стадии, определяющей скорость (RDS), в эксперименте A не будет наблюдаться значительный кинетический изотопный эффект, поскольку На общую скорость изотопное замещение не влияет. Тем не менее, этап необратимого разрыва связи CH даст первичный кинетический изотопный эффект, как и два других эксперимента, поскольку второй этап все равно будет влиять на распределение продуктов. Следовательно, в экспериментах B и C можно наблюдать кинетический изотопный эффект, даже если разрыв связи CH или CD происходит не на этапе, определяющем скорость, а на этапе, определяющем продукт.


![{\displaystyle {\frac {d[A]}{dt}}={\frac {k_{1}k_{3}[ABCD]}{k_{2}[D]+k_{3}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f71c52221df559ae3b305086cf125e2cfbfa62c4)
![{\displaystyle {\frac {d[A]}{dt}}=k_{1}[ABCD]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6cfaea2beb4c13e320a49b4f66ede1c5a85d0fba)
![{\displaystyle {\frac {d[A]}{dt}}={\frac {k_{1}k_{3}[ABCD]}{k_{2}[D]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ea50a8acf1a1307f4d9c2b4605f62578e2f79771)
соотношений констант скорости по реакциям конкуренции межмолекулярной Оценка
В конкурентных реакциях KIE рассчитывается на основе соотношений изотопного продукта или оставшихся реагентов после реакции, но эти соотношения сильно зависят от степени завершения реакции. Чаще всего изотопный субстрат состоит из молекул, меченных в определенном положении, и их немеченых обычных аналогов. [8] Также возможно в случае 13 C KIE, как и в аналогичных случаях, просто полагаться на естественное содержание изотопного углерода для экспериментов по кинетическому изотопному эффекту, устраняя необходимость в изотопной маркировке. [37] Два изотопных субстрата будут реагировать по одному и тому же механизму, но с разной скоростью. Таким образом, соотношение между количествами двух веществ в реагентах и продуктах будет постепенно меняться в ходе реакции, и это постепенное изменение можно рассматривать следующим образом: [8] Предположим, что две изотопные молекулы A 1 и A 2 вступают в необратимые реакции конкуренции следующим образом:
![{\displaystyle {\begin{aligned}{\ce {{A1}+{B}+{C}+\cdots }} \ &{\ce {->[k_{1}]P1}}\\{\ ce {{A2}+{B}+{C}+\cdots }}\ &{\ce {->[k_{2}]P2}}\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8a3e1c66994bddc0e6b39e89f23e4e0ec7a47b5c)
KIE для этого сценария оказывается следующим:

Где F 1 и F 2 относятся к доле конверсий для изотопных разновидностей A 1 и A 2 соответственно.
![{\displaystyle {\text{rate}}={-d[{\ce {A}}_{n}] \over dt}=k_{n}\times [{\ce {A}}_{n}]\times f([{\ce {B}}],[{\ce {C}}],\cdots ){\text{ where }}n=1{\text{ or }}2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/26ca6ed80abd2295998fb23de5de165477f2848e)
![{\displaystyle {1 \over k_{1}}\times {\ce {{\mathit {d}}[A1] \over [A1]}}={1 \over k_{2}}\times {\ce {{\mathit {d}}[A2] \over [A2]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ec95ce0a836f8db29e8a86da2d3a2390214f462c)
![{\displaystyle {1 \over k_{1}}\times \int \limits _{\ce {[A1]^{0}}}^{\ce {[A1]}}{d[{\ce {A}}'_{1}] \over [{\ce {A}}'_{1}]}={1 \over k_{2}}\times \int \limits _{\ce {[A2]^{0}}}^{\ce {[A2]}}{d[{\ce {A}}'_{2}] \over [{\ce {A}}'_{2}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8b3f5c1311b00398d60afa93ae27d58ac423fd32)
![{\displaystyle {k_{1} \over k_{2}}={\frac {\ce {\ln([A1]/[A1]^{0})}}{\ce {\ln([A2]/[A2]^{0})}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a4f8b9f334d8799e9e6fc51d105e53d0148086f1)


![{\displaystyle {\text{KIE}}={\frac {k_{1}}{k_{2}}}={\frac {\ln(1-F_{1})}{\ln[(1-F_{1})R/R_{0}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9f819291ca2e6641dfaf1a2d31025c40927bdd7a)
![{\displaystyle {R \over R_{0}}={\ce {{\frac {[A2]/[A1]}{[A2]^0/[A1]^0}}}}={\ce {{\frac {[A2]/[A2]^0}{[A1]/[A1]^0}}}}={\frac {1-F_{2}}{1-F_{1}}}=(1-F_{1})^{(k_{2}/k_{1})-1}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/61529d236e7c75ed167c0a1e43f996134b54b908)
Изотопное обогащение исходного материала можно рассчитать по зависимости R/R 0 от F 1 для различных КИЭ, что дает следующий рисунок. Из-за экспоненциальной зависимости даже очень низкие кинетические изотопные эффекты приводят к большим изменениям изотопного состава исходного материала при высоких конверсиях.

Если следовать продуктам, KIE можно рассчитать, используя соотношение продуктов R P вместе с R 0 следующим образом:
![{\displaystyle {k_{1} \over k_{2}}={\frac {\ln(1-F_{1})}{\ln[1-(F_{1}R_{P}/R_{0) })]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37f721659b133bfa69404286f90a8309fea92944)
Измерение кинетического изотопного эффекта естественном при изобилии
Измерение кинетического изотопного эффекта при естественном распространении — это простой общий метод измерения кинетического изотопного эффекта (КИЭ) для химических реакций , проводимых с материалами естественного содержания . Этот метод измерения KIE преодолевает многие ограничения предыдущих методов измерения KIE. Измерения KIE для меченых изотопами материалов требуют нового синтеза для каждого меченого изотопами материала (процесс часто непомерно трудный), конкурентной реакции и анализа. [5] Измерение KIE при естественном распространении позволяет избежать этих проблем, используя преимущества высокоточных количественных методов ( спектроскопия ядерного магнитного резонанса , масс-спектрометрия изотопного соотношения для выборочного измерения кинетического фракционирования изотопов ) в продукте или исходном материале для данной химической реакции .
Одноимпульсный ЯМР [ править ]
Количественная одноимпульсная спектроскопия ядерного магнитного резонанса (ЯМР) - это метод, пригодный для измерения фракционирования изотопов кинетического для измерений естественного содержания KIE. Паскаль и др. были вдохновлены исследованиями, демонстрирующими резкие различия в содержании дейтерия в идентичных соединениях из разных источников, и выдвинули гипотезу, что ЯМР можно использовать для измерения кинетических изотопных эффектов дейтерия при его естественном содержании. [39] [40] Паскаль и его коллеги проверили свою гипотезу, изучая реакцию внедрения диметилдиазомалоната в циклогексан . Паскаль и др. измерил KIE 2,2, используя 2
ЧАС
ЯМР материалов, встречающихся в природе. [40]

Синглтон и его коллеги продемонстрировали возможности 13
С
естественного содержания KIE на основе ЯМР для изучения механизма [4 + 2] циклоприсоединения изопрена Измерения с малеиновым ангидридом . [37] В предыдущих исследованиях Гаевского изотопно-обогащенных материалов наблюдались результаты KIE, которые предполагали асинхронное переходное состояние, но всегда были последовательными, в пределах ошибки, для идеально синхронного механизма реакции . [41]

Эта работа Синглтона и др. установил измерение множественных 13
С
КИЭ в рамках разработки одного эксперимента. Эти 2
ЧАС
и 13
С
Измерения KIE, определенные при естественном распространении, показали, что «внутренние» водороды диена испытывают более выраженное воздействие. 2
ЧАС
KIE, чем «внешние» водороды», а C1 и C4 испытывают значительный KIE. Эти ключевые наблюдения предполагают асинхронный циклоприсоединения изопрена механизм с реакции малеиновым ангидридом .
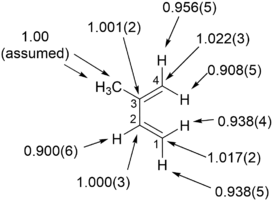
Ограничения для определения KIE при естественном содержании с помощью ЯМР заключаются в том, что извлеченный материал должен иметь подходящее количество и чистоту для анализа ЯМР (интересующий сигнал должен отличаться от других сигналов), представляющая интерес реакция должна быть необратимой, а механизм реакции не должен изменяться в течение химической реакции .
Подробности эксперимента по использованию количественного одиночного импульсного ЯМР для измерения кинетического изотопного эффекта при естественном содержании следующие: эксперимент необходимо проводить в количественных условиях, включая время релаксации 5 Тл , измеренный угол поворота 90°, цифровое разрешение не менее 5 точек на пике и сигнал:шум более 250. Необработанный FID заполняется нулями как минимум до 256 тыс. точек перед преобразованием Фурье. Спектры ЯМР фазируются, а затем обрабатываются коррекцией базовой линии нулевого порядка без какой-либо коррекции наклона. Интегрирование сигналов определяется численно с минимальным допуском для каждого интегрированного сигнала. [37] [ нужны разъяснения ]
механизма металлоорганических Примеры объяснения реакций
Коллетто и др. разработанный в региоселективном -арилирование бензо[b]тиофенов при комнатной температуре с арилйодидами в качестве партнеров сочетания и стремились понять механизм этой реакции, выполняя измерения кинетического изотопного эффекта естественного содержания с помощью одноимпульсного ЯМР. [42]




Наблюдение за первичным 13 Изотопный эффект C на C3, обратный 2 Изотопный эффект H, вторичный 13 Изотопный эффект C при C2 и отсутствие 2 Изотопный эффект H на C2; под руководством Коллетто и др. предложить механизм реакции типа Хека для региоселективного -арилирование бензо[b]тиофенов при комнатной температуре с арилйодидами в качестве партнеров сочетания. [42]
Фрост и др. стремились понять влияние добавок кислоты Льюиса на механизм энантиоселективной активации связи CN, катализируемой палладием, используя измерения кинетического изотопного эффекта естественного содержания с помощью одноимпульсного ЯМР. [43]


Первичный 13 C KIE, наблюдаемый в отсутствие BPH 3, предполагает механизм реакции с лимитирующим цис-окислением по связи C–CN цианоформамида . Добавление Bph 3 вызывает относительное снижение наблюдаемой 13 Эффект кинетического изотопа C, который привел Frost et al. предложить изменение скорости лимитирующей стадии от цис-окисления к координации палладия с цианоформамидом. [43]
DEPT-55 ЯМР [ править ]
Хотя измерения KIE при естественном распространении являются мощным инструментом для понимания механизмов реакции, количество материала, необходимое для анализа, может сделать этот метод недоступным для реакций, в которых используются дорогие реагенты или нестабильные исходные материалы. Чтобы смягчить эти ограничения, Якобсен и его коллеги разработали 1 Ч до 13 Перенос поляризации C как средство сокращения времени и материала, необходимых для измерений KIE в естественной среде обитания. Улучшение без искажений за счет переноса поляризации (DEPT) использует преимущества большего гиромагнитного отношения 1 ч закончился 13 C, чтобы теоретически улучшить чувствительность измерения в 4 раза или сократить время эксперимента в 16 раз. Этот метод измерения кинетических изотопов естественного содержания удобен для анализа реакций, содержащих нестабильные исходные материалы, а также катализаторы или продукты, которые являются относительно дорогостоящими. [44]
Якобсен и его коллеги определили катализируемое тиомочевиной гликозилирование галактозы как реакцию, которая удовлетворяет обоим вышеупомянутым критериям (дорогие материалы и нестабильные субстраты) и представляет собой реакцию с плохо изученным механизмом. [45] Гликозилирование представляет собой частный случай нуклеофильного замещения, в котором отсутствует четкое определение механистического характера S N 1 и S N 2. Присутствие кислорода рядом с местом смещения (т.е. C1) может стабилизировать положительный заряд. Эта стабилизация заряда может привести к тому, что любой потенциальный согласованный путь станет асинхронным и приблизится к промежуточным соединениям с оксокарбениевым характером механизма S N 1 гликозилирования.


Якобсен и его коллеги наблюдали небольшие нормальные KIE при C1, C2 и C5, что предполагает значительный характер оксокарбения в переходном состоянии и асинхронный механизм реакции с большой степенью разделения зарядов.
Масс- изотопного отношения спектрометрия
Высокоточная масс-спектрометрия соотношения изотопов это еще один метод измерения кинетического фракционирования изотопов (IRMS) - для измерений KIE в естественном содержании. Видлански и его коллеги продемонстрировали 34
С
естественного содержания гидролиза сульфатных моноэфиров KIE при измерении . Их наблюдение за большим KIE предполагает, что расщепление связи SO контролирует скорость и, вероятно, исключает механизм ассоциированной реакции . [46]

Основным ограничением для определения KIE при естественном распространении с использованием IRMS является необходимость селективной деградации без изотопного фракционирования до поддающихся анализу малых молекул, что является нетривиальной задачей. [37]
Тематические исследования [ править ]
водорода первичного Эффекты изотопа
KIE первичного водорода относятся к случаям, когда связь с изотопно-меченным водородом образуется или разрывается на стадии реакции, определяющей скорость и/или продукт. [5] Это наиболее часто измеряемые KIE, и большая часть ранее рассмотренной теории относится к первичным KIE. При наличии достаточных доказательств того, что перенос меченого водорода происходит на стадии, определяющей скорость реакции, если наблюдается довольно большой KIE, например, kH/kD по крайней мере 5–6 или kH/kT около 10–13 при комнатной температуре. температуры, вполне вероятно, что перенос водорода носит линейный характер и что водород достаточно симметрично расположен в переходном состоянии. Обычно невозможно комментировать туннельный вклад в наблюдаемый изотопный эффект, если только эффект не очень велик. Если первичный KIE не так велик, обычно считается, что он указывает на значительный вклад движения тяжелого атома в координату реакции, хотя это также может означать, что перенос водорода происходит по нелинейному пути. [8]
водорода вторичного Эффекты изотопа
Вторичные изотопные эффекты водорода или вторичные КИЭ (СКИЕ) возникают в тех случаях, когда изотопное замещение удалено от разрываемой связи. Тем не менее удаленный атом влияет на внутренние колебания системы, которые через изменения энергии нулевой точки (ZPE) влияют на скорости химических реакций. [47] Такие эффекты выражаются как отношения интенсивности легкого изотопа к интенсивности тяжелого изотопа и могут быть «нормальными» (отношение ≥ 1) или «обратными» (отношение <1) эффектами. [48] SKIE определяются как вторичные изотопные эффекты α, β (и т. д.), где такие префиксы относятся к положению изотопного замещения относительно реакционного центра (см. Альфа- и бета-углерод ). [49] Префикс α относится к изотопу, связанному с реакционным центром, а префикс β относится к изотопу, связанному с атомом, соседним с реакционным центром, и так далее.
В физической органической химии SKIE обсуждается с точки зрения электронных эффектов, таких как индукция, гибридизация связей или гиперконъюгация . [50] Эти свойства определяются распределением электронов и зависят от колебательно-усредненной длины связи и углов, на которые не сильно влияет изотопное замещение. Таким образом, использование термина «электронный изотопный эффект», хотя и является законным, не рекомендуется, поскольку его можно неверно истолковать, предполагая, что изотопный эффект имеет электронную, а не вибрационную природу. [49]
SKIE можно объяснить с точки зрения изменений в орбитальной гибридизации. Когда гибридизация атома углерода меняется с sp 3 sp 2 , затрагивается ряд мод колебаний (растяжения, изгиб в плоскости и вне плоскости). Изгиб в плоскости и вне плоскости в sp 3 гибридизованный углерод схож по частоте из-за симметрии sp 3 гибридизированный углерод. В СП
гибридизированный углерод, изгиб в плоскости намного жестче, чем изгиб вне плоскости, что приводит к большой разнице в частоте, ZPE и, следовательно, в SKIE (которая существует, когда существует разница в ZPE реагента и переходного состояния). ). [19] Теоретическое максимальное изменение, вызванное разницей частот изгиба, было рассчитано как 1,4. [19]
Когда углерод подвергается реакции, которая меняет его гибридизацию с sp 3 sp 2 , постоянная силы изгиба вне плоскости в переходном состоянии становится слабее по мере развития sp 2 характер и «нормальный» СКАЙ наблюдается с типичными значениями от 1,1 до 1,2. [19] И наоборот, когда гибридизация углерода меняется с sp 2 sp 3 , константы силы изгиба вне плоскости в переходном состоянии увеличиваются, и наблюдается обратный SKIE с типичными значениями от 0,8 до 0,9. [19]
В более общем смысле SKIE для обратимых реакций может быть «нормальным» в одном направлении и «обратным» в другом, если связь в переходном состоянии находится на середине жесткости между подложкой и продуктом, или они могут быть «нормальными» в обоих направлениях, если связь слабее в переходном состоянии. переходное состояние или «обратное» в обоих направлениях, если связь в переходном состоянии сильнее, чем в любом из реагентов. [48]
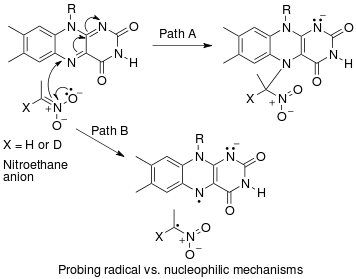
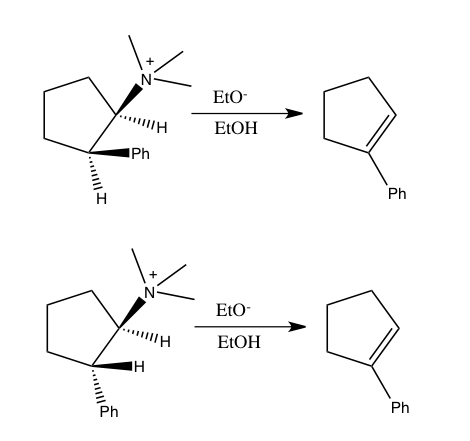
Пример «обратного» вторичного кинетического изотопного эффекта α можно увидеть в работе Фитцпатрика и Курца, которые использовали такой эффект, чтобы различить два предполагаемых пути реакции оксидазы d-аминокислот с анионами нитроалканов . [51] Путь A включает нуклеофильную атаку кофермента флавинадениндинуклеотида (FAD), тогда как путь B включает промежуточный продукт свободных радикалов. Поскольку путь A приводит к промежуточной гибридизации с изменением углерода от sp 2 sp 3 ожидается «обратное» SKIE. Если имеет место путь B, то SKIE не должно наблюдаться, поскольку промежуточный продукт свободных радикалов не изменяет гибридизацию. Было обнаружено значение SKIE 0,84 и проверен путь А, как показано на схеме ниже.
Другим примером SKIE является окисление бензиловых спиртов диметилдиоксираном, где были предложены три переходных состояния для разных механизмов. Опять же, рассматривая, как и были ли атомы водорода вовлечены в каждый из них, исследователи предсказали, будут ли они ожидать эффекта их изотопного замещения. Затем анализ экспериментальных данных реакции позволил им выбрать, какой путь наиболее вероятен, исходя из наблюдаемого изотопного эффекта. [52]
Вторичные изотопные эффекты водорода от атомов водорода метилена также были использованы, чтобы показать, что перегруппировка Коупа в 1,5-гексадиене происходит по согласованному пути перегруппировки связей, а не по одному из альтернативно предложенных путей аллильного радикала или 1,4-диила, которые все являются представлены на следующей схеме. [53]

Альтернативные механизмы перегруппировки Коупа 1,5-гексадиена: (сверху вниз), аллильный радикал, синхронно-согласованный и 1,4-диильный пути. Установлено, что преобладающим путем является средний путь, имеющий шесть делокализованных π-электронов, соответствующих ароматическому интермедиату. [53]
эффекты Стерические изотопные
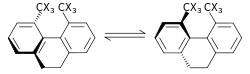 |

Эффект стерического изотопа — это SKIE, который не предполагает разрыва или образования связей. Этот эффект объясняется различной амплитудой колебаний изотопологов . [54] Примером такого эффекта является рацемизация 9,10-дигидро-4,5-диметилфенантрена. [55] Меньшая амплитуда колебаний дейтерия по сравнению с водородом в связях C–H (углерод–водород), C–D (углерод–дейтерий) приводит к меньшему радиусу Ван-дер-Ваальса или эффективному размеру в дополнение к разнице в ZPE между эти двое. Когда имеется больший эффективный объем молекул, содержащих одну, по сравнению с другой, это может проявляться в стерическом влиянии на константу скорости. В приведенном выше примере дейтерий рацемизируется быстрее, чем изотополог водорода, что приводит к стерическому изотопному эффекту. Модель стерического изотопного эффекта была разработана Бартеллом. [56] Эффект стерического изотопа обычно невелик, если только превращения не проходят через переходное состояние с серьезными стерическими препятствиями, как в процессе рацемизации, показанном выше.
Другим примером стерического изотопного эффекта является реакция проскальзывания ротаксанов. Дейтерий из-за своего меньшего эффективного размера облегчает прохождение стопоров через макроцикл, что приводит к более быстрому выведению дейтерированных ротаксанов . [57]
эффекты кинетические Обратные изотопные
Известны реакции, в которых дейтерированные соединения реагируют быстрее, чем недейтерированные, и говорят, что в этих случаях наблюдаются обратные KIE (IKIE). IKIE часто наблюдаются при восстановительном отщеплении гидридов алкилметаллов, например ( (Me 2 NCH 2 ) 2 )PtMe(H). [б] В таких случаях связь CD в переходном состоянии, представляющая собой агостическую разновидность, высоко стабилизирована по отношению к связи C–H. [58]
Обратный эффект также может возникнуть в многостадийной реакции, если общая константа скорости зависит от предварительного равновесия перед стадией, определяющей скорость , которая имеет обратный равновесный изотопный эффект . Например, скорости кислотно-катализируемых реакций обычно в 2-3 раза выше для реакций в D 2 O, катализируемых D 3 O. + чем для аналогичных реакций в H 2 O, катализируемых H 3 O + [4] : 433 Это можно объяснить механизмом специфического водородно-ионного катализа реагента R H 3 O. + (или Д 3 О + ).
- H3H3O + + Р ⇌ Правая + + Н 2 О
- относительной влажности + + Н 2 О → Н 3 О + + П
Тогда скорость образования продуктов d[P]/dt = k 2 [RH + ] знак равно k 2 K 1 [Ч 3 О + ][R] = k obs [H 3 O + ][Р]. На первом этапе H 3 O + обычно является более сильной кислотой, чем RH + . Дейтерирование смещает равновесие в сторону более прочно связанных кислотных форм RD. + в котором влияние дейтерирования на энергию нулевых колебаний больше, так что константа дейтерированного равновесия K 1D больше, чем K 1H . Этот равновесный изотопный эффект на первом этапе обычно перевешивает кинетический изотопный эффект на втором этапе, так что возникает очевидный обратный изотопный эффект и наблюдаемая общая константа скорости k obs = k 2 K 1 уменьшается. [4] : 433
растворителя водорода Кинетические эффекты изотопные
Чтобы эффекты изотопов растворителя можно было измерить, часть растворителя должна иметь другой изотопный состав, чем остальная часть. Следовательно, должны быть доступны большие количества менее распространенных видов изотопов, что ограничивает наблюдаемые изотопные эффекты растворителя изотопными заменами с участием водорода. Обнаруживаемые KIE возникают только тогда, когда растворенные вещества обменивают водород с растворителем или когда вблизи места реакции происходит специфическое взаимодействие растворенного вещества с растворителем. Оба таких явления характерны для протонных растворителей, в которых водород обменен, и они могут образовывать диполь-дипольные взаимодействия или водородные связи с полярными молекулами. [8]
углерода- изотопа Эффекты 13
Большинство органических реакций включают разрыв и образование связей с углеродом; таким образом, разумно ожидать обнаруживаемых эффектов изотопов углерода. Когда 13 В качестве метки используется C, однако изменение массы изотопа составляет всего ~ 8%, что ограничивает наблюдаемые KIE гораздо меньшими значениями, чем те, которые наблюдаются при эффектах изотопа водорода.
Компенсация изменений в 13 C естественное изобилие [ править ]
Часто самым большим источником ошибок в исследовании, которое зависит от естественного содержания углерода, является небольшое изменение естественного содержания углерода. 13 C само изобилие. Такие вариации возникают; потому что исходные материалы в реакции сами являются продуктами других реакций, которые имеют KIE и, таким образом, изотопно обогащают продукты. Чтобы компенсировать эту ошибку при использовании ЯМР-спектроскопии для определения KIE, были предложены следующие рекомендации: [38]
- Выберите углерод, удаленный от реакционного центра, который будет служить эталоном, и предположите, что он не участвует в реакции.
- В исходном материале, который не подвергся какой-либо реакции, определите отношения интегралов пиков ЯМР другого углерода к интегралам эталонного углерода.
- Получите те же соотношения для углерода в образце исходного материала после того, как он подвергся некоторой реакции.
- Отношения последних отношений к первым дают R/R 0 .
Если соблюдать эти, а также некоторые другие меры предосторожности, перечисленные Янковским, можно получить KIE с точностью до трех десятичных знаков. [38]
с элементами тяжелее углерода эффекты Изотопные
Интерпретация эффектов изотопов углерода обычно осложняется одновременным образованием и разрывом связей с углеродом. Даже реакции, которые включают только разрыв связи с углеродом, такие как реакции S N 1, предполагают усиление остальных связей с углеродом. Во многих таких реакциях эффекты изотопов уходящей группы, как правило, легче интерпретировать. Например, реакции замещения и отщепления, в которых хлор действует как уходящая группа, удобны для интерпретации, тем более что хлор действует как одноатомная разновидность без внутренних связей, усложняющих координату реакции, и имеет два стабильных изотопа: 35 кл и 37 Cl, оба с высокой численностью. Основной проблемой при интерпретации таких эффектов изотопов является сольватация уходящей группы. [8]
Из-за экспериментальных неопределенностей измерение изотопного эффекта может повлечь за собой значительную неопределенность. Часто изотопные эффекты определяются посредством дополнительных исследований ряда изотопомеров. Соответственно, весьма полезно сочетать эффекты изотопов водорода с эффектами изотопов тяжелых атомов. Например, определение изотопного эффекта азота наряду с изотопным эффектом водорода было использовано, чтобы показать, что реакция иона 2-фенилэтилтриметиламмония с этоксидом в этаноле при 40 ° C протекает по механизму E2, в отличие от альтернативных несогласованных механизмов. Этот вывод был сделан после того, как было показано, что эта реакция дает изотопный эффект азота k 14 / k 15 , равный 1,0133±0,0002, а также KIE водорода, равный 3,2 на уходящем водороде. [8]
Аналогичным образом, сочетание эффектов изотопов азота и водорода было использовано, чтобы показать, что син-элиминирование простых солей аммония также происходит по согласованному механизму, который ранее был предметом дискуссий. В следующих двух реакциях иона 2-фенилциклопентилтриметиламмония с этоксидом, обе из которых дают 1-фенилциклопентен, оба изомера проявили изотопный эффект азота k 14 / k 15 при 60 ° C. Хотя реакция транс-изомера, которая следует за син-элиминированием, имеет меньший KIE азота (1,0064), чем цис-изомер, который подвергается антиэлиминированию (1,0108); оба результата достаточно велики, чтобы указывать на ослабление связи CN в переходном состоянии, которое могло бы произойти в согласованном процессе. [с]
Другие примеры [ править ]
Поскольку кинетические изотопные эффекты возникают из-за различий в изотопных массах, наибольшие наблюдаемые КИЭ связаны с изотопными заменами водорода на дейтерий (увеличение массы в 2 раза) или тритий (увеличение массы в 3 раза). KIE из соотношений масс изотопов может достигать 36,4 с использованием мюонов. Они создали легчайший атом водорода, 0.11 H (0,113 а.е.м.), в котором электрон вращается вокруг положительного мюона (μ + ) «ядро», имеющее массу 206 электронов. Они также получили самый тяжелый атом «водорода», заменив один электрон в гелии отрицательным мюоном μ. − с образованием Heμ с атомной массой 4,116 а.е.м. Поскольку − намного тяжелее электрона, он вращается намного ближе к ядру, эффективно экранируя один протон, заставляя Heμ вести себя как 4.1 H. С этими экзотическими атомами реакция H с 1 H 2 был исследован. Константы скорости реакции самых легких и самых тяжелых аналогов водорода с 1 H 2 затем были использованы для расчета k 0,11 / k 4,1 КИЭ, в котором имеется 36,4-кратная разница в изотопных массах. В этой реакции изотопное замещение приводит к обратному кинетическому изотопному эффекту, и авторы сообщают, что KIE составляет всего 1,74 × 10. −4 , что является самым маленьким показателем KIE, когда-либо зарегистрированным. [59]
KIE приводит к определенному распределению 2 Н в природных продуктах в зависимости от пути их синтеза в природе. Таким образом, с помощью ЯМР-спектроскопии легко определить, был ли спирт в вине сброжен из глюкозы или из незаконно добавленной сахарозы .
Другой механизм реакции , который был выяснен с помощью KIE, - галогенирование толуола это : [60]
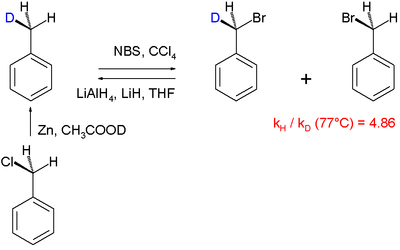
В этом конкретном «внутримолекулярном исследовании KIE» бензильный водород подвергается радикальному замещению бромом с использованием N -бромсукцинимида в качестве бромирующего агента. Было обнаружено, что PhCH 3 бромируется в 4,86 раза быстрее, чем PhCD 3 . Большой КИЕ, равный 5,56, связан с реакцией кетонов с бромом и гидроксидом натрия . [61]

В этой реакции лимитирующей стадией является образование енолята путем депротонирования кетона. В этом исследовании KIE рассчитывается на основе констант скорости реакции для обычного 2,4-диметил-3-пентанона и его дейтерированного изомера путем измерения оптической плотности .
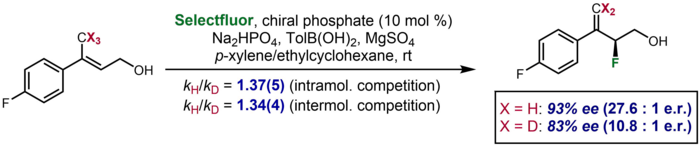
В асимметричном катализе существуют редкие случаи, когда КИЭ проявляется в виде значительной разницы в энантиоселективности, наблюдаемой для дейтерированного субстрата по сравнению с недейтерированным. Об одном примере сообщили Тосте и его коллеги, в котором дейтерированный субстрат обеспечивал энантиоселективность 83% э.и. по сравнению с 93% э.и. для недейтерированного субстрата. Эффект был использован для подтверждения дополнительных данных KIE о меж- и внутримолекулярной конкуренции, которые предполагают разрыв связи CH/D на стадии энантиоопределения. [62]
Примечания [ править ]
- ^ Это соглашение существует как для удобства номенклатуры, так и для отражения того, как эффекты кинетических изотопов дейтерия обычно изучаются экспериментально: Хотя дейтерий ( 2 H) имеет символ D, одобренный ИЮПАК, общего символа, конкретно относящегося к протию, не существует ( 1 ЧАС). Тем не менее, оказалось полезным иметь метки для обозначения соответствующих констант скорости протий- или дейтерийсодержащих изотопологов, поэтому k H и k D обычно использовались соответственно. Более того, величина KIE тогда может быть выражена как k H / k D . Эти обозначения согласуются с тем фактом, что экспериментально 2 H KIE измеряются путем сравнения скорости реакции исходного материала, обогащенного дейтерием, со скоростью реакции необогащенного исходного материала, содержащего водород в естественном количестве. Это почти всегда справедливо, поскольку на долю протия приходится ~99,9885% природного водорода, поэтому обычно нет необходимости в дальнейшем истощении дейтерия в исходном материале для получения «обогащенного протием» образца. В совокупности обозначения и экспериментальная установка привели к общей концепции дейтерия как «заместителя», который заменяет «обычный» водород в исследовании изотопного эффекта.
- ^ «Ме» означает метил , CH 3 .
- ^ На схемах ниже «Et» означает этил , C 2 H 5 ; тогда как «Ph» означает фенил , C 6 H 5 . Фенил также называют символом Φ ( фи ).
См. также [ править ]
- Кроссоверный эксперимент (химия)
- Константа равновесия # Эффект изотопного замещения
- Влияние изотопов на перекисное окисление липидов
- Кинетические изотопные эффекты RuBisCO (оксигеназы рибулозо-1,5-бисфосфаткарбоксилазы)
- Магнитный изотопный эффект
- Механизм реакции
- Переходное кинетическое фракционирование изотопов
- Уравнение Юри–Бигелайзена–Майера
Ссылки [ править ]
- ↑ Перейти обратно: Перейти обратно: а б с д и Вестэуэй КС (2006). «Использование кинетических изотопных эффектов для определения структуры переходных состояний реакций S N 2». Достижения физико-органической химии . 41 : 217–273. дои : 10.1016/S0065-3160(06)41004-2 . ISBN 978-0-12-033541-1 .
- ^ Линн К.Р., Янквич PE (5 августа 1961 г.). «Изотопное фракционирование по метиловому углероду в реакциях цианид-иона с метилхлоридом и метилбромидом». Журнал Американского химического общества . 83 (15): 3220–3223. дои : 10.1021/ja01476a012 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж Аткинс П., де Паула Дж (2006). Физическая химия Аткинса (8-е изд.). Издательство Оксфордского университета . стр. 286–288 , 816–818. ISBN 978-0-19-870072-2 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час Лейдлер К.Дж. (1987). Химическая кинетика (3-е изд.). Харпер и Роу. ISBN 978-0-06-043862-3 .
- ↑ Перейти обратно: Перейти обратно: а б с д Симмонс Э.М., Хартвиг Дж.Ф. (март 2012 г.). «К интерпретации кинетических изотопных эффектов дейтерия в функционализации связи C – H комплексами переходных металлов». Angewandte Chemie, международное издание . 51 (1): 3066–72. дои : 10.1002/anie.201107334 . ПМИД 22392731 .
- ^ Пуарье Р.А., Ван Ю, Вестэуэй К.С. (март 1994 г.). «Теоретическое исследование связи между кинетическими изотопными эффектами вторичного альфа-дейтерия и структурой переходных состояний S N 2». Журнал Американского химического общества . 116 (6): 2526–2533. дои : 10.1021/ja00085a037 .
- ↑ Перейти обратно: Перейти обратно: а б с д и Бансель Э., Ли CC (1977). Изотопы в катионных реакциях . Изотопы в органической химии. Том. 5. Амстердам: Эльзевир. ISBN 978-0-444-41927-9 . OCLC 867217247 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я Меландер Л., Сондерс WH (1980). Скорости реакций изотопных молекул . Нью-Йорк: Уайли.
- ^ Бигелайзен Дж., Вольфсберг М. (январь 1957 г.). «Теоретические и экспериментальные аспекты изотопных эффектов в химической кинетике». Достижения химической физики . 1 : 15–76.
- ^ Если мюоний (μ + и – ) рассматривается как изотоп водорода, то в принципе возможны и более крупные КИЭ. Однако исследования мюония ограничены коротким периодом полураспада мюона (22 микросекунды) (см. Вилья Х, Корчадо Х.К., Гонсалес-Лафон А., Ллуч Х.М., Трулар Д.Г. (ноябрь 1998 г.). «Объяснение кинетических изотопных эффектов дейтерия и мюония при присоединении атома водорода к олефину». Журнал Американского химического общества . 120 (46): 12141–2. дои : 10.1021/ja982616i . на примере изотопного эффекта k Mu / k H. )
- ^ Бигелейзен Дж. (август 1949 г.). «Относительные скорости реакции изотопных молекул». Журнал химической физики . 17 (8): 675–678. Бибкод : 1949ЖЧФ..17..675Б . дои : 10.1063/1.1747368 .
- ↑ Перейти обратно: Перейти обратно: а б с Лоури Т.Х., Ричардсон К.С. (1987). Механизм и теория органической химии (3-е изд.). Нью-Йорк: Харпер и Роу. стр. 256 . ISBN 978-0-06-044084-8 . OCLC 14214254 .
- ^ Плотник Б.К. (1984). Определение механизмов органических реакций . Нью-Йорк: Уайли. п. 86. ИСБН 978-0-471-89369-1 . ОСЛК 9894996 .
- ^ Плотник Б.К. (февраль 2010 г.). «Кинетические изотопные эффекты: открытие нетрадиционного». Природная химия . 2 (2): 80–2. Бибкод : 2010НатЧ...2...80С . дои : 10.1038/nchem.531 . ПМИД 21124393 .
- ^ Кэрролл Ф.А. (2010). Взгляды на структуру и механизм органической химии (2-е изд.). Хобокен, Нью-Джерси: Джон Уайли. ISBN 978-0-470-27610-5 . OCLC 286483846 .
- ^ Кварт Х (1 декабря 1982 г.). «Температурная зависимость первичного кинетического эффекта изотопа водорода как механистический критерий». Отчеты о химических исследованиях . 15 (12): 401–408. дои : 10.1021/ar00084a004 . ISSN 0001-4842 .
- ^ Стрейтвизер А., Ягов Р.Х., Фэйи Р.К., Сузуки С. (май 1958 г.). «Кинетические изотопные эффекты при ацетолизе дейтерированных циклопентилтозилатов 1, 2». Журнал Американского химического общества . 80 (9): 2326–32. дои : 10.1021/ja01542a075 .
- ^ Суэйн К.Г., Стиверс Э.К., Ройвер-младший Дж.Ф., Шаад Л.Дж. (1 ноября 1958 г.). «Использование эффектов изотопа водорода для идентификации атакующего нуклеофила при енолизации кетонов, катализируемой уксусной кислотой». Журнал Американского химического общества . 80 (21): 5885–5893. дои : 10.1021/ja01554a077 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж Анслин Э.В., Догерти Д.А. (2006). Современная физико-органическая химия . Университетские научные книги. стр. 428–437 . ISBN 978-1-891389-31-3 .
- ^ Разауй М (2003). Квантовая теория туннелирования . Всемирная научная . ISBN 978-981-238-019-7 .
- ^ Силбей Р.Дж., Альберти Р.А., Бавенди М.Г. (2005). Физическая химия . Джон Уайли и сыновья . стр. 326–338. ISBN 978-0-471-21504-2 .
- ^ Боргис Д., Хайнс Дж.Т. (1993). «Динамическая теория скорости туннельного переноса протонов в растворе: общая формулировка». Химическая физика . 170 (3): 315–346. Бибкод : 1993CP....170..315B . дои : 10.1016/0301-0104(93)85117-Q .
- ↑ Перейти обратно: Перейти обратно: а б Кришталик Л.И. (май 2000 г.). «Механизм переноса протона: краткое описание» . Biochimica et Biophysica Acta (BBA) — Биоэнергетика . 1458 (1): 6–27. дои : 10.1016/S0005-2728(00)00057-8 . ПМИД 10812022 .
- ^ Зуев П.С., Шеридан Р.С., Албу Т.В., Трухлар Д.Г., Хроват Д.А., Борден В.Т. (февраль 2003 г.). «Туннелирование углерода из одного квантового состояния». Наука . 299 (5608): 867–70. Бибкод : 2003Sci...299..867Z . дои : 10.1126/science.1079294 . ПМИД 12574623 . S2CID 20959068 .
- ^ Фудзисаки Н., Руф А., Гауманн Т. (1987). «Туннельные эффекты в реакциях переноса атома водорода, изученные с помощью температурной зависимости кинетических эффектов изотопа водорода и дейтерия». Журнал физической химии . 91 (6): 1602–1606. дои : 10.1021/j100290a062 .
- ^ Льюис Э.С., Фундерберк Л. (1967). «Скорость и изотопные эффекты при переносе протона от 2-нитропропана к пиридиновым основаниям». Журнал Американского химического общества . 89 (10): 2322–2327. дои : 10.1021/ja00986a013 .
- ^ Дьюар М.Дж., Хили Э.Ф., Руис Дж.М. (1988). «Механизм 1,5-сигматропного водородного сдвига в 1,3-пентадиене». Журнал Американского химического общества . 110 (8): 2666–2667. дои : 10.1021/ja00216a060 .
- ^ фон Деринг В., Чжао X (июль 2006 г.). «Влияние дейтерия на кинетику 1,5-водородного сдвига цисоид-замкнутого 1,3(Z)-пентадиена, 2-метил-10-метиленбицикло[4.4.0]дец-1-ена: доказательства туннелирования? ". Журнал Американского химического общества . 128 (28): 9080–5. дои : 10.1021/ja057377v . ПМИД 16834382 .
- ^ В этом исследовании KIE измеряется с помощью чувствительного протонного ЯМР . Экстраполированный KIE при 25 °C составляет 16,6, но погрешность высока.
- ^ Коэн А., Клинман Дж. П. (июль 1999 г.). «Водородное туннелирование в биологии» . Химия и биология . 6 (7): Р191-8. дои : 10.1016/S1074-5521(99)80058-1 . ПМИД 10381408 .
- ^ Уайльд Т.К., Блотни Дж., Поллак Р.М. (май 2008 г.). «Экспериментальные доказательства усиленного ферментами связанного движения/квантово-механического туннелирования водорода с помощью кетостероид-изомеразы». Журнал Американского химического общества . 130 (20): 6577–85. дои : 10.1021/ja0732330 . ПМИД 18426205 .
- ^ Трулар Д.Г., Гао Х., Альгамбра К., Гарсиа-Вилока М., Корчадо Х., Санчес М., Вилла Х. (2002). «Учет квантовых эффектов в моделировании кинетики ферментов». Отчеты о химических исследованиях . 35 (6): 341–349. дои : 10.1021/ar0100226 . ПМИД 12069618 .
- ^ Коэн, А; Клинман, Дж. П. (1998). «Ферментный катализ: за пределами классических парадигм». Отчеты о химических исследованиях . 31 (7): 397–404. дои : 10.1021/ar9701225 .
- ^ Мэгги Ф., Райли У.Дж. (2010). «Математическая обработка изотопологического и изотопомерного видообразования и фракционирования в биохимической кинетике» . Geochimica et Cosmochimica Acta . 74 (6): 1823. Бибкод : 2010GeCoA..74.1823M . дои : 10.1016/j.gca.2009.12.021 . S2CID 55422661 .
- ^ Ньюолл, А. Рэймонд; Хейс, Джон; Бетелл, Дональд (1 января 1974 г.). «Промежуточные продукты разложения алифатических диазосоединений. Часть XI. Механистические исследования реакции дифенилметилена с аминами в растворе». Журнал Химического общества, Perkin Transactions 2 (11): 1307–1312. дои : 10.1039/P29740001307 . ISSN 1364-5471 .
- ^ Бансель Э., Ли CC (1977). Углерод-13 в органической химии . Изотопы в органической химии. Том. 3. Амстердам: Эльзевир. ISBN 978-0-444-41472-4 . OCLC 606113159 .
- ↑ Перейти обратно: Перейти обратно: а б с д Синглтон Д.А., Томас А.А. (сентябрь 1995 г.). «Высокоточное одновременное определение множественных малых кинетических изотопных эффектов при естественном изотопе». Журнал Американского химического общества . 117 (36): 9357–9358. дои : 10.1021/ja00141a030 .
- ↑ Перейти обратно: Перейти обратно: а б с Янковский С. (январь 2009 г.). «Применение ЯМР-спектроскопии в исследованиях изотопных эффектов». Годовые отчеты по ЯМР-спектроскопии . 68 : 149–191. дои : 10.1016/S0066-4103(09)06803-3 . ISBN 978-0-12-381041-0 .
- ^ Мартин Дж.Дж., Мартин М.Л. (1984). «Метка дейтерием на естественном уровне содержания, изученная с помощью количественного 2H ЯМР в сильном поле». Буквы тетраэдра . 22 (36): 3525–3528. дои : 10.1016/s0040-4039(01)81948-1 .
- ↑ Перейти обратно: Перейти обратно: а б Паскаль-младший Р.А., Баум М.В., Вагнер К.К., Роджерс Л.Р. (сентябрь 1984 г.). «Измерение кинетических изотопных эффектов дейтерия в органических реакциях методом ЯМР-спектроскопии естественного содержания дейтерия». Журнал Американского химического общества . 106 (18): 5377–5378. дои : 10.1021/ja00330a071 .
- ^ Гаевски Дж. Дж., Петерсон К.Б., Кагель Дж. Р., Хуанг Ю. Дж. (декабрь 1989 г.). «Изменение структуры переходного состояния в реакции Дильса-Альдера из-за эффектов вторичного кинетического изотопа дейтерия. Реакция почти симметричных диенов и диенофилов почти синхронна». Журнал Американского химического общества . 111 (25): 9078–9081. дои : 10.1021/ja00207a013 .
- ↑ Перейти обратно: Перейти обратно: а б Коллетто С., Ислам С., Хулиа-Эрнандес Ф., Ларроса I (февраль 2016 г.). «Прямое β-арилирование тиофена и бензо[b]тиофена при комнатной температуре и кинетические доказательства пути типа Хека» . Журнал Американского химического общества . 138 (5): 1677–83. дои : 10.1021/jacs.5b12242 . ПМЦ 4774971 . ПМИД 26788885 .
- ↑ Перейти обратно: Перейти обратно: а б Фрост ГБ, Серраторе Н.А., Огилви Дж.М., Дуглас СиДжей (апрель 2017 г.). «Механистическая модель энантиоселективного внутримолекулярного цианоамидирования алкенов посредством катализируемой палладием активации связи C-CN» . Журнал органической химии . 82 (7): 3721–3726. дои : 10.1021/acs.joc.7b00196 . ПМЦ 5535300 . ПМИД 28294618 .
- ^ Кван Э.Э., Пак Ю., Бессер Х.А., Андерсон Т.Л., Якобсен Э.Н. (январь 2017 г.). «Измерения кинетического изотопного эффекта 13C с помощью переноса поляризации» . Журнал Американского химического общества . 139 (1): 43–46. дои : 10.1021/jacs.6b10621 . ПМК 5674980 . ПМИД 28005341 .
- ^ Пак Ю, Харпер К.К., Куль Н., Кван Э.Э., Лю Р.Ю., Якобсен Э.Н. (январь 2017 г.). «Макроциклические бис-тиомочевины катализируют реакции стереоспецифического гликозилирования» . Наука . 355 (6321): 162–166. Бибкод : 2017Sci...355..162P . doi : 10.1126/science.aal1875 . ПМЦ 5671764 . ПМИД 28082586 .
- ^ Берлинхэм Б.Т., Пратт Л.М., Дэвидсон Э.Р., Шайнер В.Дж., Фонг Дж., Видлански Т.С. (октябрь 2003 г.). «Влияние изотопа 34S на гидролиз сульфатного эфира: механистические последствия». Журнал Американского химического общества . 125 (43): 13036–7. дои : 10.1021/ja0279747 . ПМИД 14570471 .
- ^ Хенниг С., Освальд Р.Б., Шматц С. (март 2006 г.). «Вторичный кинетический изотопный эффект при нуклеофильном замещении: квантово-механический подход». Журнал физической химии А. 110 (9): 3071–9. Бибкод : 2006JPCA..110.3071H . дои : 10.1021/jp0540151 . ПМИД 16509628 .
- ↑ Перейти обратно: Перейти обратно: а б Клеланд WW (декабрь 2003 г.). «Использование изотопных эффектов для определения ферментативных механизмов» . Журнал биологической химии . 278 (52): 51975–84. дои : 10.1074/jbc.X300005200 . ПМИД 14583616 .
- ↑ Перейти обратно: Перейти обратно: а б ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Онлайн исправленная версия: (2006–) « Вторичный изотопный эффект ». дои : 10.1351/goldbook.S05523
- ^ «Определение изотопного эффекта вторичного» . Химический словарь . Химитул.
- ^ Курц К.А., Фитцпатрик П.Ф. (1997). «РН и вторичные кинетические изотопные эффекты на реакцию оксидазы D-аминокислот с нитроалкановыми анионами: доказательства прямой атаки на флавин карбанионами». Журнал Американского химического общества . 119 (5): 1155–1156. дои : 10.1021/ja962783n .
- ^ Анжелис Ю.С., Хацакис Н.С., Смону И., Орфанопулос М. (2006). «Окисление бензиловых спиртов диметилдиоксираном. Вопрос о согласованных и ступенчатых механизмах, исследованных с помощью кинетических изотопных эффектов». Буквы тетраэдра . 42 (22): 3753–3756. дои : 10.1016/S0040-4039(01)00539-1 .
- ↑ Перейти обратно: Перейти обратно: а б Хоук К.Н., Густавсон С.М., Блэк К.А. (октябрь 1992 г.). «Теоретические вторичные кинетические изотопные эффекты и интерпретация геометрии переходного состояния. 1. Перегруппировка Коупа». Журнал Американского химического общества . 114 (22): 8565–72. дои : 10.1021/ja00048a032 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Стерический изотопный эффект ». doi : 10.1351/goldbook.S06001
- ^ Мислоу К. , Грейв Р., Гордон А.Дж., Валь Г.Х. (1963). «Заметка о стерических изотопных эффектах. Конформационные кинетические изотопные эффекты при рацемизации 9,10-дигидро-4,5-диметилфенантрена». Журнал Американского химического общества . 85 (8): 1199–1200. дои : 10.1021/ja00891a038 .
- ^ Бартелл Л.С. (1 сентября 1961 г.). «Роль несвязанных отталкиваний во вторичных изотопных эффектах. I. Эффекты альфа- и бета-замещения.1». Журнал Американского химического общества . 83 (17): 3567–3571. дои : 10.1021/ja01478a006 .
- ^ Фелдер Т., Шалли, Калифорния (май 2003 г.). «Вторичные изотопные эффекты на реакцию скольжения ротаксанов: высокоточное измерение стерического размера». Ангеванде Хеми . 42 (20): 2258–60. дои : 10.1002/anie.200350903 . ПМИД 12772156 .
- ^ Черчилль, Дэвид Г.; Джанак, Кевин Э.; Виттенберг, Джошуа С.; Паркин, Джерард (11 января 2003 г.). «Нормальные и обратные первичные кинетические эффекты изотопа дейтерия для восстановительного удаления связи C-H и реакций окислительного присоединения молибденоценовых и вольфрамоценовых комплексов: данные о промежуточных соединениях σ-комплекса бензола» . Дж. Ам. хим. Соц . 125 (5): 1403–1420 . Проверено 26 января 2023 г.
- ^ Флеминг Д.Г., Арсено Д.Д., Сухоруков О., Брюэр Дж.Х., Мильке С.Л., Шац Г.К., Гарретт Б.С., Петерсон К.А., Трулар Д.Г. (январь 2011 г.). «Кинетические изотопные эффекты для реакций мюонного гелия и мюония с H2». Наука . 331 (6016): 448–50. Бибкод : 2011Sci...331..448F . дои : 10.1126/science.1199421 . ПМИД 21273484 . S2CID 206530683 .
- ^ Виберг КБ, Слаф Л.Х. (1958). «Эффект изотопа дейтерия при галогенировании боковой цепи толуола». Журнал Американского химического общества . 80 (12): 3033–3039. дои : 10.1021/ja01545a034 .
- ^ Линч Р.А., Винченти С.П., Лин Ю.Т., Смакер Л.Д., Субба Рао С.К. (1972). «Аномальное кинетическое влияние изотопов водорода на скорость ионизации некоторых диалкилзамещенных кетонов». Журнал Американского химического общества . 94 (24): 8351–8356. дои : 10.1021/ja00779a012 .
- ^ Цзы В, Ван Ю.М., Тосте Ф.Д. (сентябрь 2014 г.). «Групповая стратегия управления хиральным анионным фазовым переносом аллильных спиртов in situ» . Журнал Американского химического общества . 136 (37): 12864–7. дои : 10.1021/ja507468u . ПМК 4183625 . ПМИД 25203796 .
Дальнейшее чтение [ править ]
- Белл Р.П., Крукс Дж.Э. (20 июля 1965 г.). «Кинетические эффекты изотопа водорода при ионизации некоторых кетоновых веществ». Труды Лондонского королевского общества А. 286 (1406): 285–299. Бибкод : 1965RSPSA.286..285B . дои : 10.1098/rspa.1965.0144 . S2CID 96761478 .