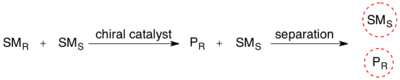
Кинетическое разрешение
В органической химии кинетическое разрешение является средством дифференциации двух энантиомеров в рацемической смеси . При кинетическом разрешении два энантиомера реагируют с разной скоростью реакции в химической реакции с хиральным катализатором или реагентом, в результате чего образуется энантиообогащенный образец менее реакционноспособного энантиомера. [ 1 ] В отличие от хирального разрешения , кинетическое разделение зависит не от различных физических свойств диастереомерных продуктов, а скорее от различных химических свойств рацемических исходных материалов. Энантиомерный избыток (э.и.) непрореагировавшего исходного материала постоянно возрастает по мере образования большего количества продукта, достигая 100% непосредственно перед полным завершением реакции. Кинетическое разрешение зависит от различий в реакционной способности между энантиомерами или энантиомерными комплексами.
Кинетическое разрешение можно использовать для получения хиральных молекул в органическом синтезе . Реакции кинетического разделения с использованием чисто синтетических реагентов и катализаторов гораздо менее распространены, чем использование ферментативного кинетического разделения в применении к органическому синтезу, хотя за последние 30 лет был разработан ряд полезных синтетических методов. [ 2 ]
История
[ редактировать ]Первое зарегистрированное кинетическое разрешение было достигнуто Луи Пастером . После реакции водного раствора рацемического тартрата аммония с плесенью Penicillium glaucum он повторно выделил оставшийся тартрат и обнаружил, что он обладает левовращающим действием . [ 3 ] Хиральные микроорганизмы, присутствующие в плесени, избирательно катализировали метаболизм ( R , R )-тартрата, оставляя избыток ( S , S )-тартрата.
О кинетическом разрешении синтетическими методами впервые сообщили Марквальд и Маккензи в 1899 году при этерификации рацемической миндальной кислоты оптически активным (-)- ментолом . При наличии избытка рацемической кислоты они наблюдали, что образование сложного эфира, полученного из (+)- миндальной кислоты, происходит быстрее, чем образование сложного эфира из (-)-миндальной кислоты. Было обнаружено, что непрореагировавшая кислота имела небольшой избыток (-)-миндальной кислоты, а позже было показано, что сложный эфир дает (+)-миндальную кислоту при омылении. Важность этого наблюдения заключалась в том, что теоретически, если бы использовалась половина эквивалента (-)-ментола, можно было бы получить высокоэнантиообогащенный образец (-)-миндальной кислоты. Это наблюдение привело к успешному кинетическому разрешению других хиральных кислот, что положило начало использованию кинетического разрешения в органической химии. [ 4 ] [ 5 ]

Теория
[ редактировать ]Кинетическое разрешение - возможный метод необратимой дифференциации пары энантиомеров из-за (потенциально) разных энергий активации. Хотя оба энантиомера по определению находятся на одном и том же уровне свободной энергии Гиббса , и продукты реакции с обоими энантиомерами также находятся на равных уровнях, , или энергия переходного состояния , может различаться. На изображении ниже энантиомер R имеет более низкую и, таким образом, будет реагировать быстрее, чем энантиомер S.


Идеальным кинетическим разрешением является такое, при котором реагирует только один энантиомер, т.е. k R >>k S . Селективность kS (s) кинетического разрешения связана с скорости реакции энантиомеров R и S, kR и kS соответственно , соотношением s=kR / , константами для kR > kS . Эту селективность можно также назвать относительной скоростью реакции . Это можно записать через разность свободной энергии между переходными состояниями с высокой и низкой энергией: . [ 6 ]


Селективность также можно выразить через ee извлеченного исходного материала и конверсию (c), если предположить кинетику первого порядка (в субстрате). Если предположить, что S-энантиомер рацемата исходного материала будет выделен в избытке, можно выразить концентрации (мольные доли) S- и R-энантиомеров как
![{\displaystyle [S]={\frac {(1+ee)(1-c)}{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bedb0ada89fbba54020c618907425dc858a2591b)
![{\displaystyle [R]={\frac {(1-ee)(1-c)}{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1a61e8391de522b72a3580bd1214d5112057d741)
где ee – ee исходного материала. Обратите внимание, что при c=0, что означает начало реакции, , где они означают начальные концентрации энантиомеров. Тогда для стехиометрического хирального разделяющего агента B*:

![{\displaystyle {\frac {d[S]}{dt}}=-k_{S}[S][B^{*}]\ подразумевает \log[S]=-k_{S}[B^{* }]t+\log S_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0cb65f6ee0307b80ab311cb9d09726947bd97ac1)
Обратите внимание, что если разделяющий агент является стехиометрическим и ахиральным, с хиральным катализатором, член [B*] не появляется. Тем не менее, используя аналогичное выражение для R, мы можем выразить s как
![{\displaystyle s={\frac {k_{R}}{k_{S}}}={\frac {\log[R]-\log R_{0}}{\log[S]-\log S_{ 0}}}={\frac {\log[(1-c)(1-ee)]+\log {\frac {1}{2}}-\log R_{0}}{\log[(1-c)(1+ee)]+\log {\frac {1}{2}}-\log S_{0}}}={\frac {\log[ (1-c)(1-ee)]}{\log[(1-c)(1+ee)]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/156b26edecc36af36374614253bb2842be3e550b)
Если мы хотим выразить это через энантиомерный избыток продукта ee", мы должны воспользоваться тем фактом, что для продуктов R' и S' из R и S соответственно
![{\displaystyle ee''={\frac {[R']-[S']}{[R']+[S']}}={\frac {ee(1-c)}{c}}\ подразумевает ee=ee''{\frac {c}{1-c}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0876f7366b97d07d9a002cb750fdb1a42ca25448)
Отсюда мы видим, что


что дает нам


которые, когда мы подключаем наше выражение для s, полученное выше, дают
![{\displaystyle s = {\frac {\log[1-c(1+ee'')]}{\log[1-c(1-ee'')]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6d142162d7ab054c445d0ee122e6ed2b2880103a)
Коэффициент конверсии ( c ) и селективности ( s ) могут быть выражены только через энантиомерные избытки исходного материала и продукта (ee и ee'' соответственно):
![{\displaystyle c={\frac {ee}{ee+ee''}}\подразумевает s={\frac {\log[(ee''-ee\cdot ee'')/(ee+ee'') ]}{\log[(ee''+ee\cdot ee'')/(ee+ee'')]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/882e4515e695067b8c4864f5a5adf0e984064631)
Кроме того, выражения для c и ee можно параметризовать, чтобы получить явные выражения для C и ee через t. Во-первых, явное решение для [S] и [R] как функций от t дает
![{\displaystyle {\frac {d[S]}{dt}}=-k_{S}[S]\implies S={\frac {1}{2}}e^{-k_{S}t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8fcf79858e21ac5c20f56338e038c1a28e2b6e77)
что, подставленное в выражения для ee и c, дает
![{\displaystyle ee={\frac {[S]-[R]}{[S]+[R]}}={\frac {e^{-k_{S}t}-e^{-k_{R }t}}{e^{-k_{S}t}+e^{-k_{R}t}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/537dcb1d65d95eea71ade3ed04fb39937e993fb2)
![{\displaystyle c=1-{\big (}[S]-[R]{\big)}=1- {\frac {e^{-k_{S}t}+e^{-k_{R} т}}{2}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/002346d2f288670c618bfcddfabc48db5fe6a46b)
Без ограничения общности можно допустить k S =1, что дает k R =s, упрощая приведенные выше выражения. Аналогичным образом можно вывести выражение для ee″ как функции t.

Таким образом, графики зависимости ee и ee″ от c могут быть построены с использованием t в качестве параметра и различных значений s, образующих разные кривые, как показано ниже.
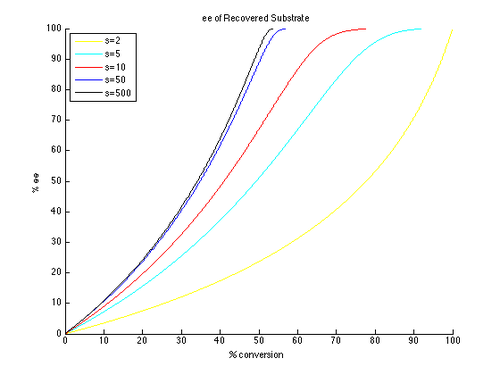
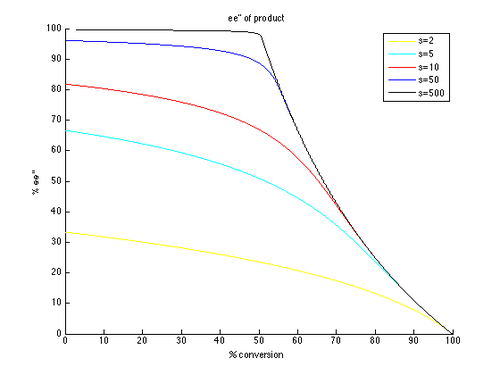
Как можно видеть, высокие энантиомерные избытки гораздо легче достижимы для непрореагировавшего исходного материала. Однако существует компромисс между ee и конверсией: более высокая ee (восстановленного субстрата) достигается при более высокой конверсии и, следовательно, при более низком выходе изолированного продукта. Например, при коэффициенте селективности всего 10 можно получить 99% э.и. при конверсии примерно 70%, что дает выход около 30%. Напротив, чтобы получить хорошие показатели эффективности и выхода продукта, необходимы очень высокие коэффициенты селективности. Например, при коэффициенте селективности 10 недостижимо значение ee″ выше примерно 80%, а для более реалистичных преобразований получаются значительно более низкие значения ee″. Для получения высокоэнантиообогащенного продукта с разумным выходом требуется селективность более 50.
Это упрощенная версия истинной кинетики кинетического разрешения. Предположение о том, что реакция имеет первый порядок по субстрату, является ограничивающим, и возможно, что зависимость от субстрата может зависеть от конверсии, что приводит к гораздо более сложной картине. В результате общий подход состоит в том, чтобы измерять и сообщать только выходы и ee, поскольку формула для k rel применима только к идеализированному кинетическому разрешению. Легко предположить, что образуется первоначальный комплекс субстрат-катализатор, который может свести на нет кинетику первого порядка. Тем не менее, сделанные общие выводы по-прежнему полезны для понимания влияния селективности и конверсии на ee.
Практичность
[ редактировать ]С появлением асимметричного катализа необходимо рассмотреть целесообразность использования кинетического разрешения для получения энантиочистых продуктов. Даже для продукта, который может быть получен асимметричным каталитическим или вспомогательным путем, рацемат может быть значительно дешевле, чем энантиочистый материал, что приводит к повышению экономической эффективности даже с неизбежной «потерей» 50% материала. Следующие условия были предложены в качестве необходимых условий для практического кинетического разрешения: [ 6 ]
- недорогой рацемат и катализатор
- невозможен подходящий энантиоселективный, хиральный пул или классический путь разрешения.
- разделение происходит выборочно при низких загрузках катализатора
- разделение исходного материала и продукта легко
На сегодняшний день разработан ряд катализаторов кинетического разделения, которые удовлетворяют большинству, если не всем, вышеуказанным критериям, что делает их весьма практичными для использования в органическом синтезе. В следующих разделах будет обсуждаться ряд ключевых примеров.
Реакции с использованием синтетических реагентов
[ редактировать ]Реакции ацилирования
[ редактировать ]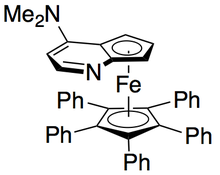
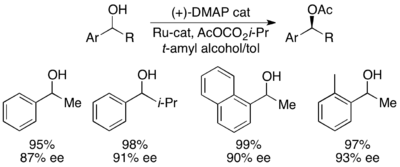
Грегори Фу и его коллеги разработали методологию, использующую хиральный аналог DMAP для достижения превосходного кинетического разрешения вторичных спиртов. [ 7 ] Первоначальные исследования с использованием эфира в качестве растворителя, низких загрузок катализатора (2 моль%), уксусного ангидрида в качестве ацилирующего агента и триэтиламина при комнатной температуре дали селективность в диапазоне 14-52, что соответствует ee восстановленного спиртового продукта до 99,2%. . [ 8 ] Однако скрининг растворителей показал, что использование трет-амилового спирта увеличивает как реакционную способность, так и селективность. [ 9 ]

Для эталонного субстрата 1-фенилэтанола это соответствовало 99% э.и. непрореагировавшего спирта при конверсии 55% при работе при 0 °C. Эта система оказалась подходящей для разделения ряда арилалкилкарбинолов с селективностью до 95 и низким содержанием катализатора 1%, как показано ниже, с использованием (-)-энантиомера катализатора. Это привело к получению сильно энантиообогащенных спиртов при очень низких конверсиях, что также дало отличные выходы. Кроме того, высокая селективность приводит к получению высокоэнантиообогащенных ацилированных продуктов с 90% ее образца ацилированного спирта для о-толилметилкарбинола с s = 71.
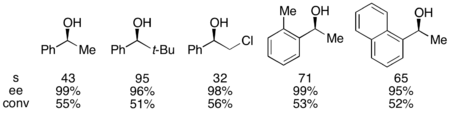
Кроме того, Фу сообщил о первом высокоселективном ацилировании рацемических диолов (а также о десимметризации мезодиолов). При низкой загрузке катализатора (1%) энантиообогащенный диол был извлечен с выходом 98% и 43%, а диацетат - с выходом 39% и 99% ее. Оставшуюся часть материала извлекали в виде смеси моноацетата.

Также было показано, что планарно-хиральный катализатор DMAP эффективен при кинетическом растворении пропаргиловых спиртов. [ 10 ] Однако в этом случае селективность оказалась самой высокой без присутствия какого-либо основания. При работе с 1 мол.% катализатора при 0°C можно достичь селективности до 20. Ограничения этого метода включают требование наличия ненасыщенной функциональной группы, такой как карбонил или алкены, в удаленном алкинильном положении. Спирты, разделенные с использованием (+)-энантиомера катализатора DMAP, показаны ниже.

Фу также продемонстрировал способность своего хирального катализатора DMAP расщеплять аллильные спирты. [ 11 ] Эффективная селективность зависела от присутствия геминального или цис-заместителя в спиртсодержащей группе, за заметным исключением транс-фенилового спирта, который проявлял самую высокую селективность. Используя 1-2,5 мол% (+)-энантиомера катализатора DMAP, приведенные ниже спирты разделяли в присутствии триэтиламина.


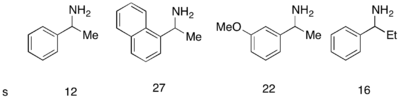
Хотя аналоговый катализатор Фу DMAP исключительно хорошо работал для кинетического разделения рацемических спиртов, его не удалось использовать для кинетического разделения аминов. Был разработан аналогичный катализатор PPY*, который при использовании с новым ацилирующим агентом позволил успешно проводить ацилирование аминов с кинетическим разрешением. При использовании 10 мол% (-)-PPY* в хлороформе при –50 °C при ацилировании аминов наблюдалась селективность от хорошей до очень хорошей, как показано ниже. [ 12 ] Аналогичный протокол был разработан для кинетического разрешения индолинов. [ 13 ]
Эпоксидирование и дигидроксилирование
[ редактировать ], Эпоксидирование Шарплесса разработанное К. Барри Шарплессом в 1980 году. [ 14 ] был использован для кинетического разделения рацемической смеси аллильных спиртов. [ 15 ] [ 16 ] Несмотря на то, что этот метод чрезвычайно эффективен при растворении ряда аллильных спиртов, он имеет ряд недостатков. Время реакции может достигать 6 дней, и катализатор не подлежит вторичной переработке. Однако кинетическое разрешение асимметричного эпоксидирования Шарплесса остается одним из наиболее эффективных синтетических кинетических разрешений на сегодняшний день. В качестве катализатора можно использовать ряд различных тартратов; Типичная схема показана ниже с использованием диизопропилтартрата . Этот метод нашел широкое применение для ряда вторичных аллильных спиртов. [ 17 ]

Асимметричное дигидроксилирование Шарплесса также использовалось в качестве метода кинетического разрешения. [ 18 ] [ 19 ] Однако этот метод не получил широкого распространения, поскольку одно и то же разрешение можно достичь разными, более экономичными способами. Кроме того, было показано, что эпоксидирование Ши влияет на кинетическое разрешение ограниченного набора олефинов. [ 20 ] Этот метод также не получил широкого распространения, но представляет механистический интерес.
Эпоксидные отверстия
[ редактировать ]
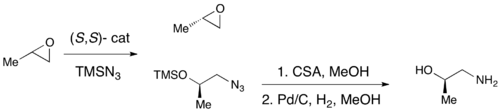
Хотя энантиоселективное эпоксидирование было успешно достигнуто с использованием эпоксидирования по Шарплесу, эпоксидирования по Ши и эпоксидирования по Якобсену , ни один из этих методов не позволяет обеспечить эффективный асимметричный синтез концевых эпоксидов, которые являются ключевыми хиральными строительными блоками. Из-за дешевизны большинства рацемических концевых эпоксидов и их невозможности подвергнуть классическому разделению, эффективное кинетическое разделение концевых эпоксидов может служить очень важной синтетической методологией. В 1996 году Якобсен и его коллеги разработали методологию кинетического разделения эпоксидов посредством раскрытия нуклеофильного кольца с атакой азид-аниона. Показан катализатор (R,R). [ 21 ] Катализатор может эффективно, при таких низких нагрузках, как 0,5 мол.%, энантиоселективно открывать эпоксид в концевом положении, давая энантиообогащенный исходный эпоксид и 1,2-азидоспирты. Выходы почти количественные, а ee были превосходными (≥95% почти во всех случаях). 1,2-азидоспирты можно гидрировать с получением 1,2-аминоспиртов, как показано ниже.

В 1997 году группа Якобсена опубликовала методологию, которая улучшила их более раннюю работу, позволяя использовать воду в качестве нуклеофила в раскрытии эпоксида. При использовании почти идентичного катализатора наблюдалось значение ee, превышающее 98% как для извлеченного исходного эпоксида, так и для продукта 1,2-диола. В приведенном ниже примере гидролитическое кинетическое разделение (HKR) проводилось в масштабе 58 граммов, в результате чего было получено 26 г (44%) энантиоризованного эпоксида в >99% ее и 38 г (50%) диола в 98% эээ. [ 22 ]

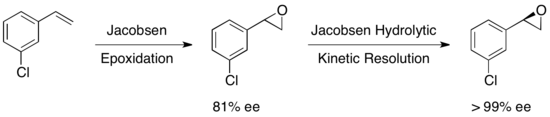
Было исследовано множество других субстратов, при этом выходы извлеченного эпоксида варьировались от 36 до 48% при >99% ее. Гидролитическое кинетическое разделение по Якобсену можно использовать в сочетании с эпоксидированием по Якобсену для получения энантиочистых эпоксидов из некоторых олефинов, как показано ниже. Первое эпоксидирование дает слегка энантиообогащенный эпоксид, а последующее кинетическое разделение дает по существу один энантиомер. Преимуществом этого подхода является возможность уменьшить количество гидролитического расщепления, необходимое для достижения высокой энантиоселективности, что позволяет достичь общего выхода примерно до 90% в пересчете на олефин. [ 23 ]
В конечном счете, кинетическое разрешение раскрытия эпоксида Якобсена обеспечивает высокую энантиомерную чистоту эпоксида и продукта в условиях отсутствия растворителя или с низким содержанием растворителя и применяется в больших масштабах. Методика Якобсена для HKR, в частности, чрезвычайно привлекательна, поскольку ее можно осуществлять в многотонном масштабе и использовать воду в качестве нуклеофила, что приводит к чрезвычайно экономически эффективным промышленным процессам. Несмотря на впечатляющие достижения, HKR обычно применяется для разделения простых концевых эпоксидов с одним стереоцентром. Совсем недавно Д.А. Деваланкар с соавт. сообщили об элегантном протоколе, включающем двухстереоцентрированный сокатализируемый HKR рацемических концевых эпоксидов, несущих соседние заместители, связывающие C – C. [ 24 ]
Окисления
[ редактировать ]
Рёдзи Нойори и его коллеги разработали методологию кинетического разделения бензильных и аллильных вторичных спиртов посредством трансферного гидрирования. Комплекс рутения катализирует окисление более реакционноспособного энантиомера из ацетона с образованием непрореагировавшего энантиочистого спирта, окисленного кетона и изопропанола. В примере, проиллюстрированном ниже, воздействие на 1-фенилэтанол (S,S)-энантиомером катализатора в присутствии ацетона приводит к выходу 51%, 94% ее (R)-1-фенилэтанола, а также 49% ацетофенона. и изопропанол в качестве побочного продукта. [ 25 ]
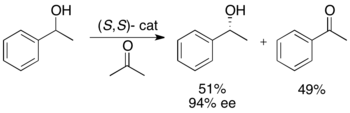
Эта методология по сути является противоположностью асимметричного переноса гидрирования кетонов Нойори. [ 26 ] которые путем восстановления дают энантиообогащенные спирты. Это ограничивает привлекательность метода кинетического разрешения, поскольку существует аналогичный метод получения тех же продуктов без потери половины материала. Таким образом, кинетическое разрешение будет осуществляться только в том случае, если рацемический спирт будет как минимум вдвое дешевле кетона или будет значительно более доступен.

Кроме того, Уэмура и Хидаи разработали рутениевый катализатор для кинетического расщепления окисления бензиловых спиртов, позволяющий получать высокоэнантиообогащенные спирты с хорошими выходами. [ 27 ] Комплекс может, как и катализатор Нойори, влиять на перенос гидрирования между кетоном и изопропанолом с образованием энантиообогащенного спирта, а также влиять на кинетическое разрешение рацемического спирта, давая энантиочистый спирт (>99% э.и.) и окисленный кетон с ацетоном в качестве побочного продукта. . Он очень эффективен при энантиоселективном восстановлении кетонов, давая большинство бензиловых спиртов с э.и. >99%, и может расщеплять ряд рацемических бензиловых спиртов с получением высоких выходов (до 49%) одиночных энантиомеров, как показано ниже. Этот метод имеет те же недостатки, что и кинетическое разрешение Нойори, а именно то, что доступ к спиртам также можно получить путем энантиоселективного восстановления кетонов. Кроме того, сообщалось только об одном энантиомере катализатора.
гидрирование
[ редактировать ]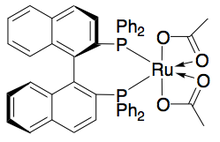
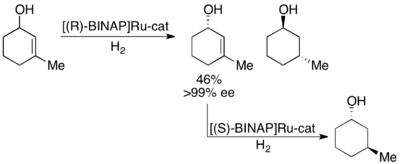
Нойори также продемонстрировал кинетическое разделение аллильных спиртов путем асимметричного гидрирования олефина. [ 28 ] С помощью комплекса Ru[BINAP] селективное гидрирование может дать высокие значения ee ненасыщенного спирта в дополнение к гидрированному спирту, как показано ниже. Таким образом, второе гидрирование оставшегося энантиообогащенного аллилового спирта даст энантиомерно чистые образцы обоих энантиомеров насыщенного спирта. Нойори разделил ряд аллильных спиртов с выходами от хороших до отличных и от хороших до отличных (до >99%).
Метатезис замыкания кольца
[ редактировать ]
Ховейда и Шрок разработали катализатор для кинетического разделения метатезиса с замыканием цикла диенилаллиловых спиртов. [ 29 ] Молибденалкилиденовый катализатор избирательно катализирует один энантиомер для осуществления метатезиса с замыканием кольца, в результате чего образуется энантиочистый спирт и энантиочистое замкнутое кольцо, как показано ниже. Катализатор наиболее эффективен при расщеплении 1,6-диенов. Однако небольшие структурные изменения в субстрате, такие как увеличение межалкенового расстояния до 1,7, иногда могут вызвать необходимость использования другого катализатора, что снижает эффективность этого метода.

Ферментативные реакции
[ редактировать ]Ацилирование
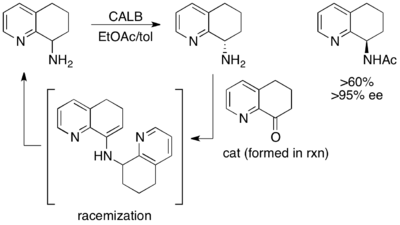
[ редактировать ]Как и в случае с процедурами синтетического кинетического разрешения, кинетическое разрешение ферментативного ацилирования нашло самое широкое применение в синтетическом контексте. Особенно важным было использование ферментативного кинетического разрешения для эффективного и дешевого получения аминокислот. В коммерческом масштабе метод Дегусса с использованием ацилаз способен расщеплять многочисленные природные и неприродные аминокислоты. Рацемические смеси могут быть приготовлены посредством синтеза Стрекера , а использование ацилазы почек свиньи (для субстратов с прямой цепью) или фермента плесени Aspergillus oryzae (для субстратов с разветвленной боковой цепью) может эффективно давать энантиообогащенные аминокислоты с высоким (85-) Выход 90%). Непрореагировавший исходный материал может быть рацемизирован in situ, что обеспечивает динамическое кинетическое разрешение. [ 30 ]

Кроме того, липазы широко используются для кинетического разрешения как в академических, так и в промышленных условиях. [ 31 ] [ 32 ] Липазы использовались для разделения первичных спиртов, вторичных спиртов, ограниченного числа третичных спиртов, карбоновых кислот, диолов и даже хиральных алленов. Липаза из Pseudomonas cepacia (PSL) наиболее широко используется для разделения первичных спиртов и использовалась вместе с винилацетатом в качестве ацилирующего агента для кинетического разделения первичных спиртов, показанных ниже.
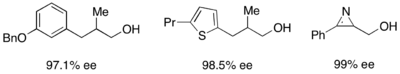
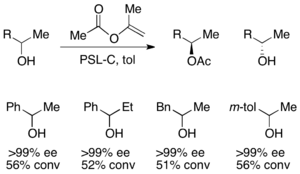
Для разделения вторичных спиртов эффективно использовалась липаза pseudomonas cepecia (PSL-C) для получения превосходных ee ( R )-энантиомера спирта. [ 33 ] Использование изопропенилацетата в качестве ацилирующего агента приводит к образованию ацетона в качестве побочного продукта, который эффективно удаляется из реакции с помощью молекулярных сит .
Окисления и восстановления
[ редактировать ]Пекарские дрожжи (BY) использовались для кинетического разделения α-стереогенных карбонильных соединений. [ 34 ] [ 35 ] Фермент избирательно восстанавливает один энантиомер, образуя сильно энантиообогащенный спирт и кетон, как показано ниже.

Пекарские дрожжи также использовались для кинетического разделения вторичных бензиловых спиртов путем окисления. [ 36 ] Хотя сообщалось об отличной эффективности извлеченного спирта, обычно для них требуется конверсия >60%, что приводит к снижению выхода. Пекарские дрожжи также использовались для кинетического разрешения путем восстановления β-кетоэфиров. [ 37 ] Однако, учитывая успех Нойори в разрешении тех же подложек, подробно описанный далее в этой статье, это не нашло особого применения.
Динамическое кинетическое разрешение
[ редактировать ]Динамическое кинетическое разрешение (DKR) происходит, когда рацемат исходного материала способен легко эпимеризоваться, что приводит к образованию по существу рацемической смеси исходного материала на всех этапах реакции. Тогда теоретически может образоваться энантиомер с более низким барьером активации с выходом до 100%. Это контрастирует со стандартным кинетическим разрешением, которое обязательно имеет максимальный выход 50%. По этой причине динамическое кинетическое разрешение имеет чрезвычайно практическое применение в органическом синтезе. Наблюдаемая динамика основана на принципе Кертина-Хэммета . Барьер реакции любого энантиомера обязательно выше, чем барьер эпимеризации, в результате чего образуется кинетическая яма, содержащая рацемат. Это эквивалентно записи: для k R >k S ,


Был опубликован ряд отличных обзоров, последний раз в 2008 году, в которых подробно описывается теория и практическое применение DKR. [ 38 ] [ 39 ] [ 40 ]
Асимметричное гидрирование Нойори
[ редактировать ]является Асимметричное гидрирование кетонов Нойори прекрасным примером динамического кинетического разрешения в действии. Энантиомерные β-кетоэфиры могут подвергаться эпимеризации , и выбор хирального катализатора, обычно в форме Ru[(R)-BINAP]X 2 , где X представляет собой галоген , приводит к тому, что один из энантиомеров реагирует преимущественно быстрее. Относительная свободная энергия типичной реакции показана ниже. [ 41 ] [ 42 ] Как можно видеть, свободная энергия промежуточного продукта эпимеризации ниже, чем у переходных состояний гидрирования, что приводит к быстрой рацемизации и высоким выходам одного энантиомера продукта.
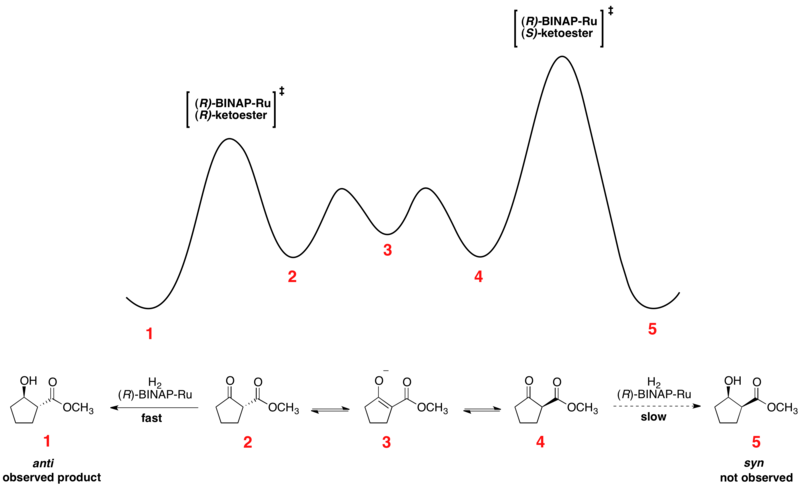
Энантиомеры взаимопревращаются посредством общего енола , который представляет собой энергетический минимум, расположенный между энантиомерами. Показанная реакция дает 93% образца анти- продукта, показанного выше. Выбор растворителя, по-видимому, оказывает большое влияние на диастереоселективность, поскольку и дихлорметан , и метанол проявляют эффективность для определенных субстратов. Нойори и другие также разработали новые катализаторы, которые улучшили как ее, так и диастереомерное соотношение (dr).

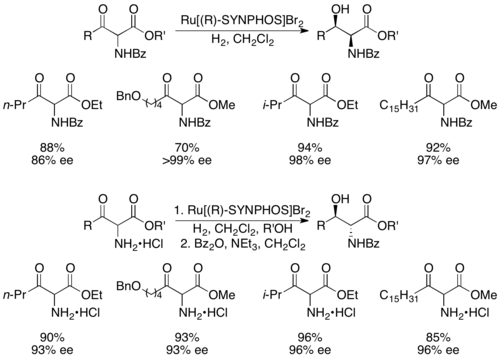
Жене и его коллеги разработали SYNPHOS , аналог BINAP, который образует комплексы рутения, которые осуществляют высокоселективное асимметричное гидрирование. [ 43 ] Было показано, что энантиочистый Ru[SYNPHOS]Br 2 селективно гидрирует рацемические α-амино-β-кетоэфиры до энантиочистых аминоспиртов, как показано ниже, с использованием (R)-SYNPHOS. [ 44 ] 1,2- синаминоспирты получали из бензоилзащищенных аминосоединений, а антипродукты - из гидрохлоридных солей амина.
Модификация фу-ацилирования
[ редактировать ]
Недавно Грегори Фу и его коллеги сообщили об модификации своей предыдущей работы по кинетическому разрешению для получения эффективного динамического кинетического разрешения. [ 45 ] Используя рутениевый катализатор рацемизации, показанный справа, и свой планарный хиральный катализатор DMAP, Фу продемонстрировал динамическое кинетическое разрешение вторичных спиртов с выходом до 99% и 93% ее, как показано ниже. Продолжается работа по дальнейшему развитию применения широко используемого катализатора DMAP для динамического кинетического разрешения.
Ферментативное динамическое кинетическое разрешение
[ редактировать ]Сообщалось о ряде ферментативных динамических кинетических разрешений. [ 46 ] Яркий пример использования PSL эффективно расщепляет рацемические ацилоины в присутствии триэтиламина и винилацетата в качестве ацилирующего агента. [ 47 ] Как показано ниже, продукт был выделен с выходом 75% и э.и. 97%. Без присутствия основания происходило регулярное кинетическое разделение, что приводило к выходу 45% ацилированного продукта с >99% ее и 53% исходного материала с 92% ее.
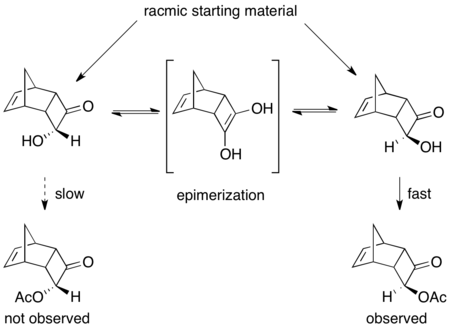
Еще одним прекрасным, хотя и не очень продуктивным примером является кинетическое разрешение (±)-8-амино-5,6,7,8-тетрагидрохинолина. При воздействии липазы B Candida antarctica (CALB) в толуоле и этилацетате в течение 3–24 часов происходит нормальное кинетическое разрешение, что приводит к выходу 45% (97% ее исходного материала) и выходу 45% (>97% ее ацилированного амина). . Однако, если реакции перемешиваться в течение 40–48 часов, восстанавливается рацемический исходный материал и >60% из >95% ее ацилированного продукта. [ 48 ]
Здесь непрореагировавший исходный материал рацемизируется in situ через димерный енамин, что приводит к извлечению более 50% продукта энантиочистого ацилированного амина.
Хемоферментативное динамическое кинетическое разрешение
[ редактировать ]Сообщалось о ряде процедур, в которых используются преимущества химического реагента/катализатора для проведения рацемизации исходного материала и фермента для избирательной реакции с одним энантиомером, что называется химиоферментативным динамическим кинетическим разрешением. [ 49 ] PSL-C использовали вместе с рутениевым катализатором (для рацемизации) для получения энантиочистых (>95% ее) δ-гидроксилактонов. [ 50 ]

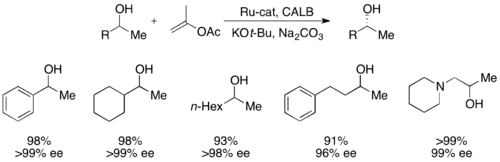
Совсем недавно Бэквалль разделил вторичные спирты с выходами до 99% и ee до >99% с использованием CALB и комплекса рацемизации рутения. [ 51 ]
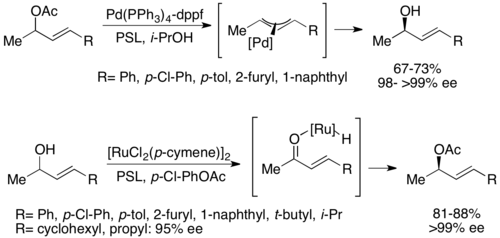
Второй тип химиоферментативного динамического кинетического разрешения включает образование π-аллильного комплекса аллилацетата с палладием . Здесь рацемизация происходит с потерей ацетата, образуя катионный комплекс с центром переходного металла, как показано ниже. [ 52 ] Было показано, что палладий облегчает эту реакцию, а рутений влияет на аналогичную реакцию, также показанную ниже. [ 53 ]
Параллельное кинетическое разрешение
[ редактировать ]При параллельном кинетическом разрешении (PKR) рацемическая смесь реагирует с образованием двух неэнантиомерных продуктов, часто по совершенно разным путям реакции. При использовании PKR нет компромисса между конверсией и ее, поскольку образующиеся продукты не являются энантиомерами. [ 54 ] [ 55 ] Одна из стратегий PKR заключается в удалении менее реакционноспособного энантиомера (по направлению к желаемому хиральному катализатору) из реакционной смеси путем воздействия на него второго набора условий реакции, которые предпочтительно взаимодействуют с ним, в идеале с примерно равной скоростью реакции. Таким образом, оба энантиомера расходуются разными путями с одинаковой скоростью. Эксперименты PKR могут быть стереодивергентными, региодивергентными или структурно дивергентными. [ 56 ] Один из наиболее эффективных PKR, зарегистрированных на сегодняшний день, был осуществлен Ёсито Киши в 1998 году; CBS привело к стереоселективному восстановлению с образованием двух диастереомеров с э.и. >99%, как показано ниже. Восстановление рацемического стероидного кетона [ 57 ]

ПКР также осуществляли с использованием ферментных катализаторов. С помощью гриба Mortierella isabellina NRRL 1757 восстановление рацемических β-кетонитрилов дает два диастереомера, которые можно разделить и повторно окислить с получением высокоэнантиочистых β-кетонитрилов. [ 58 ] Однако весьма полезные с синтетической точки зрения параллельные кинетические разрешения еще предстоит открыть. Был открыт ряд процедур, которые дают приемлемые значения эриэли и выходов, но очень мало примеров, которые дают высокоселективное параллельное кинетическое разрешение, а не просто несколько селективные реакции. Например, параллельное кинетическое разделение 4-алкиналов Фу дает очень энантиообогащенный циклобутанон с низким выходом и слегка энантиообогащенный циклопентенон, как показано ниже. [ 59 ]

Теоретически параллельное кинетическое разрешение может дать наивысшую эффективность продуктов, поскольку только один энантиомер дает каждый желаемый продукт. Например, для двух дополнительных реакций, обе с s = 49, 100% конверсия даст продукты с выходом 50% и 96% э.и. Для этих же значений потребуется s=200 для простого кинетического разрешения. Таким образом, перспективы PKR продолжают привлекать большое внимание. Сокращение Киши CBS остается одним из немногих примеров выполнения этого обещания.
См. также
[ редактировать ]- Хиральные вспомогательные вещества
- Синтез хирального пула
- Хиральное разрешение
- Энантиоселективный синтез
Ссылки
[ редактировать ]- ^ Фио, JC; Каган, HB (1988). «Кинетическое разрешение». В Элиэле, Эль; Вилен, С.Х. (ред.). Темы стереохимии . Том. 18. Нью-Йорк: John Wiley and Sons, Inc., стр. 249–340.
- ^ Робинсон, ДЕЖЕ; Булл, SD (2005). «Стратегии кинетического разрешения с использованием неферментативных катализаторов». Тетраэдр: Асимметрия . 14 (11): 1407–1446. дои : 10.1016/S0957-4166(03)00209-X .
- ^ Пастер, LC (1858). «Воспоминание о брожении винной кислоты». ЧР акад. наук. Париж . 46 :615–618.
- ^ Марквальд, В.; Маккензи, А. (1899). «О принципиально новом методе расщепления рацемических соединений на активные компоненты» . Бер. Немецкий. Хим . 32 (2): 2130–2136. дои : 10.1002/cber.189903202130 .
- ^ Роджер, Р.; Рид, Дж. (1952). «Александр Маккензи: 1869-1951» . Некрологи членов Королевского общества . 8 (21): 206–228. дои : 10.1098/rsbm.1952.0014 .
- ^ Jump up to: а б Кейт, Дж. М.; Ларроу, Дж. Ф.; Якобсен, Э.Н. (2001). «Практические соображения по реакциям кинетического разрешения». Адв. Синтез. Катал . 343 : 5–26. doi : 10.1002/1615-4169(20010129)343:1<5::AID-ADSC5>3.0.CO;2-I .
- ^ Вурц, Р.П.; Ли, ЕС; Рубль, ЮК; Фу, GC (2007). «Синтез и разделение планарно-хиральных производных 4-(диметиламино)пиридина». Адв. Синтез. Катал . 349 (14–15): 2345–2352. дои : 10.1002/adsc.200700219 .
- ^ Рубль, ЮК; Латам, штат Ха; Фу, GC (1997). «Эффективное кинетическое разрешение вторичных спиртов плоскохиральным аналогом 4-(диметиламино)пиридина. Использование группы Fe(C 5 Ph 5 ) в асимметричном катализе». Дж. Ам. хим. Соц . 119 (6): 1492–1493. дои : 10.1021/ja963835b .
- ^ Рубль, ЮК; Тведделл, Дж.; Фу, GC (1998). «Кинетическое разделение арилакилкарбинолов, катализируемое планарно-хиральным производным DMAP: новый эталон неферментативного ацилирования». Дж. Орг. Хим . 63 (9): 2794–2795. дои : 10.1021/jo980183w .
- ^ Тао, Б.; Рубль, ЮК; Хойк, Д.А.; Фу, GC (1999). «Неферментативное кинетическое разрешение пропаргиловых спиртов с помощью планарно-хирального производного DMAP: кристаллографическая характеристика ацилированного катализатора». Дж. Ам. хим. Соц . 121 (21): 2091–5092. дои : 10.1021/ja9906958 .
- ^ Бельмен-Лапонназ, С.; Тведделл, Дж.; Рубль, ЮК; Брейтлинг, FM; Фу, GC (2000). «Кинетическое разделение аллильных спиртов с помощью неферментативного катализатора ацилирования; применение к синтезу натуральных продуктов». хим. Коммун. (12): 2091–5092. дои : 10.1039/B002041I .
- ^ Арай, С.; Бельмен-Лапонназ, С.; Фу, GC (2001). «Кинетическое разделение аминов с помощью неферментативного катализатора ацилирования». Энджью. хим. Межд. Эд . 133 (1): 240–242. doi : 10.1002/1521-3757(20010105)113:1<240::AID-ANGE240>3.0.CO;2-E .
- ^ Арп, ФО; Фу, GC (2006). «Кинетическое разрешение инолинов с помощью неферментативного катализатора ацилирования» . Дж. Ам. хим. Соц . 128 (44): 14264–14265. дои : 10.1021/ja0657859 . ПМК 2569996 . ПМИД 17076493 .
- ^ Кацуки, Т.; Шарплесс, КБ (1980). «Первый практический метод асимметричного эпоксидирования». Дж. Ам. хим. Соц . 102 (18): 5974–5976. дои : 10.1021/ja00538a077 .
- ^ Мартин, В.; Вудард, С.; Кацуки, Т.; Ямада, Ю.; Икеда, М.; Шарплесс, КБ (1981). «Кинетическое разделение рацемических аллильных спиртов путем энантиоселективного эпоксидирования. Путь к веществам абсолютной энантиомерной чистоты?». Дж. Ам. хим. Соц . 103 (23): 6237–6240. дои : 10.1021/ja00410a053 .
- ^ Гао, Юн; Кландер, Дж. М.; Хэнсон, РМ; Масамунэ, Х.; Ко, С.Ю.; Шарплесс, КБ (1987). «Каталитическое асимметричное эпоксидирование и кинетическое разрешение: модифицированные процедуры, включая дериватизацию in situ». Дж. Ам. хим. Соц . 109 (19): 5765–5780. дои : 10.1021/ja00253a032 .
- ^ Китано, Ю.; Мацумото, Т.; Сато, Ф. (1988). «Высокоэффективное кинетическое разделение γ- и β-триметилсилил вторичных аллильных спиртов путем безострого асимметричного эпоксидирования». Тетраэдр . 44 (13): 4073–4086. дои : 10.1016/S0040-4020(01)86657-6 .
- ^ ВанНьевензе, М.С.; Шарплесс, КБ (1993). «Кинетическое разделение рацемических олефинов посредством асимметричного дигидроксилирования». Дж. Ам. хим. Соц . 115 (17): 7864–7865. дои : 10.1021/ja00070a037 .
- ^ Кори, Э.Дж.; Ноэ, MC; Гузман-Перес, А. (1995). «Кинетическое разрешение путем энантиоселективного дигидроксилирования вторичных аллиловых эфиров 4-метоксибензоата с использованием механически разработанного катализатора на основе алкалоида хинного дерева». Дж. Ам. хим. Соц . 117 (44): 10817–10824. дои : 10.1021/ja00149a004 .
- ^ Лоренц, Дж. К.; Фрон, М.; Чжоу, X.; Чжан, младший; Тан, Ю.; Берк, К.; Ши, Ю. (2005). «Исследование переходного состояния асимметричного эпоксидирования, опосредованного диоксираном, посредством кинетического разрешения и десимметризации». Дж. Орг. Хим . 70 (8): 2904–2911. дои : 10.1021/jo048217p . ПМИД 15822948 .
- ^ Ларроу, Дж. Ф.; Шаус, SE; Якобсен, Э.Н. (1996). «Кинетическое разрешение концевых эпоксидов посредством высокорегиоселективного и энантиоселективного раскрытия кольца с помощью TMSN3. Эффективный каталитический путь к 1,2-аминоспиртам». Дж. Ам. хим. Соц . 118 (31): 7420–7421. дои : 10.1021/ja961708+ .
- ^ Токунага, М.; Ларроу, Дж. Ф.; Какиучи, Ф.; Якобсен, Э.Н. (1997). «Асимметричный катализ с водой: эффективное кинетическое разделение концевых эпоксидов посредством каталитического гидролиза». Наука . 277 (5328): 936–938. дои : 10.1126/science.277.5328.936 . ПМИД 9252321 .
- ^ Брандес, Б.Д.; Якобсен, Э.Н. (1997). «Синтез энантиочистого 3-хлорстиролоксида с помощью асимметричной последовательности эпоксидирования-гидролитического кинетического разрешения». Тет. Асимм . 8 (23): 3927–3933. дои : 10.1016/S0957-4166(97)00568-5 .
- ^ Судалай, А.; Карабал, ПО; Деваланкар, Д.А. (2013). «Оптически чистые γ-бутиролактоны и эпоксидные сложные эфиры через два стереоцентрированных HKR 3-замещенных эпоксидных сложных эфиров: формальный синтез (-)-пароксетина, Ro 67-8867 и (+)-эльданолида». Орг. Биомол. Хим . 11 (8): 1280–1285. дои : 10.1039/c3ob27321k . ПМИД 23334653 .
- ^ Хасигути, С.; Фуджи, А.; Хаак, К.-Дж.; Мацумура, К.; Икария, Т.; Ноёри, Р. (1997). «Кинетическое разделение рацемических вторичных спиртов путем RuII-катализируемого переноса водорода». Энджью. хим. Межд. Эд . 36 (3): 288–290. дои : 10.1002/anie.199702881 .
- ^ Хасигути, С.; Фуджи, А.; Такехара, Дж.; Икария, Т.; Ноёри, Р. (1995). «Асимметричное трансферное гидрирование ароматических кетонов, катализируемое хиральными комплексами рутения (II)». Дж. Ам. хим. Соц . 117 (28): 7562–7563. дои : 10.1021/ja00133a037 .
- ^ Нишибаяси, Ю.; Такей, И.; Уэмура, С.; Хидай, М. (1999). «Чрезвычайно высокая энантиоселективная окислительно-восстановительная реакция кетонов и спиртов, катализируемая RuCl2 (PPh3) (оксазолинилферроценилфосфин)». Металлоорганические соединения . 18 (12): 2291–2293. дои : 10.1021/om990210o .
- ^ Китамура, М.; Касахара, И.; Манабе, К.; Ноёри, Р.; Такая, Х. (1988). «Кинетическое разделение рацемических аллильных спиртов путем гидрирования, катализируемого BINAP-рутением (II)». Дж. Орг. Хим . 53 (3): 708–710. дои : 10.1021/jo00238a048 .
- ^ Ховейда, АХ; Шрок, Р.Р. (2001). «Каталитический асимметричный метатезис олефинов». хим. Евро. Дж . 7 (5): 945–950. doi : 10.1002/1521-3765(20010302)7:5<945::AID-CHEM945>3.0.CO;2-3 . ПМИД 11303874 .
- ^ Патент США 6656710 , Bommarius & Verseck, «Способ производства аминокислот с использованием рацемазы и ацилата», передан Degussa AG.
- ^ Ганем, А.; Абул-Энейн, HY (2005). «Применение липаз для кинетического разделения рацематов». Хиральность . 17 (1): 1–15. дои : 10.1002/чир.20089 . ПМИД 15515046 .
- ^ «Хиральная технология: промышленный биокатализ стандартными гидролитическими ферментами». Журнал специальной химии . 27 (8): 38. 2007.
- ^ Ганем, А.; Шуриг, В. (2003). «Необратимая переэтерификация вторичных спиртов, катализируемая липазой, с использованием изопропенилацетата» (PDF) . Ежемесячные журналы по химии . 134 (8): 1151–1157. дои : 10.1007/s00706-003-0025-1 . S2CID 96922176 .
- ^ Сантаньелло, Э.; Феррабоски, П.; Гризенти, П.; Манзокки, А. (1992). «Биокаталитический подход к получению энантиомерно чистых хиральных строительных блоков». хим. Преподобный . 92 (5): 1071–1140. дои : 10.1021/cr00013a016 .
- ^ Тикоцци, К.; Занаротти, Антонио (1989). «Энантиоселективное микробное восстановление 5-ацетилизоксазолинов - новый метод стереохимического контроля снижения дрожжей». Либигс Анн. Хим . 1989 (12): 1257–1259. дои : 10.1002/jlac.198919890299 .
- ^ Фантен, Г.; Фоганьоло, М.; Медичи, А.; Педрини, П.; Поли, С. (1993). «Кинетическое разделение 1-арил- и 1-гетероарилэтанолов путем окисления пекарскими дрожжами». Тетраэдр Летт . 34 (5): 883–884. дои : 10.1016/0040-4039(93)89039-S . hdl : 11392/462444 .
- ^ Брукс, Д.В.; Уилсон, М.; Уэбб, М. (1987). «Различные ферментативные реакции энантиомерной пары: одновременное двойное кинетическое разделение кетоэфира пекарскими дрожжами». Дж. Орг. Хим . 52 (11): 2244–2248. дои : 10.1021/jo00387a026 .
- ^ Пеллиссье, Х. (2008). «Последние разработки в области динамического кинетического разрешения». Тетраэдр . 64 (8): 1563–1601. дои : 10.1016/j.tet.2007.10.080 .
- ^ Пеллиссье, Х. (2003). «Динамическое кинетическое разрешение». Тетраэдр . 59 (42): 8291–8327. дои : 10.1016/S0040-4020(03)01022-6 .
- ^ Уорд, РС (1995). «Динамическое кинетическое разрешение». Тетраэдр: Асимметрия . 6 (7): 1475–1490. дои : 10.1016/0957-4166(95)00179-S .
- ^ Китамура, М.; Токунага, М.; Ноёри, Р. (1993). «Количественное выражение динамического кинетического разрешения хирально-лабильных энантиомеров: стереоселективное гидрирование 2-замещенных эфиров 3-оксокарбоновых кислот, катализируемое комплексами BINAP-рутений (II)». Дж. Ам. хим. Соц . 115 (1): 144–152. дои : 10.1021/ja00054a020 .
- ^ Ноёри, Р.; Икеда, Т.; Окума, Т.; Видхальм, М.; Китамура, М.; Такая, Х.; Акутагава, С.; Сайо, Н.; Сайто, Т. (1989). «Стереоселективное гидрирование посредством динамического кинетического разрешения». Дж. Ам. хим. Соц . 111 (25): 9134–9135. дои : 10.1021/ja00207a038 .
- ^ де Поль, SD; Желен, С.; Ратовеломанана-Видал, В.; Женет, Япония; Чемпион, Н.; Деллис, П. (2003). «Исследования по синтезу и молекулярному моделированию SYNPHOS®, нового эффективного дифосфанового лиганда для асимметричного гидрирования, катализируемого рутением». Евро. Дж. Орг. Хим . 2003 (10): 1931–1941. дои : 10.1002/ejoc.200200634 .
- ^ Мордант, К.; Ратовеломанана-Видал, В.; Дюнкельманн, П.; Жене, Ж.-П. (2004). «Универсальный путь получения син- и анти-α-амино-β-гидроксиэфиров из β-кетоэфиров с помощью динамического кинетического разрешения с использованием катализатора Ru-SYNPHOS®». Евро. Дж. Орг. Хим . 2004 (14): 3017–3026. дои : 10.1002/ejoc.200400078 .
- ^ Ли, С.Ю.; Мерфи, Дж. М.; Укай, А.; Фу, GC (2012). «Неферментативное динамическое кинетическое разделение вторичных спиртов посредством энантиоселективного ацилирования: синтетические и механистические исследования» . Дж. Ам. хим. Соц . 134 (36): 15149–15153. дои : 10.1021/ja307425g . ПМЦ 3447740 . ПМИД 22934603 .
- ^ Пеллиссье, Х. (2003). «Динамическая переэтерификация трициклического ацилоина, имеющего латентную мезоструктуру, опосредованная липазой-триэтиламином: новый путь к оптически чистому оксодициклопентадиену». Тетраэдр . 59 (42): 8291–9327. дои : 10.1016/S0040-4020(03)01022-6 .
- ^ Танигучи, Т.; Огасавара, К. (1997). «Динамическая переэтерификация трициклического ацилоина, имеющего латентную мезоструктуру, опосредованная липазой-триэтиламином: новый путь к оптически чистому оксодициклопентадиену». Химические коммуникации (15): 1399–1400. дои : 10.1039/A702910A .
- ^ Кроуфорд, Дж. Б.; Скерль, RT; Бриджер, Дж.Дж. (2007). «Спонтанное ферментативно-опосредованное динамическое кинетическое разрешение 8-амино-5,6,7,8-тетрагидрохинолина». Дж. Орг. Хим . 72 (2): 669–671. дои : 10.1021/jo062037t . ПМИД 17221995 .
- ^ Памиес, О.; Беквалль, Ж.-Э. (2004). «Хемоферментативное динамическое кинетическое разрешение». Тенденции в биотехнологии . 22 (3): 130–135. дои : 10.1016/j.tibtech.2004.01.005 . ПМИД 15036863 .
- ^ Памиес, О.; Беквалль, Ж.-Э. (2002). «Ферментативное кинетическое разрешение и химиоферментативное динамическое кинетическое разрешение δ-гидроксиэфиров. Эффективный путь к хиральным δ-лактонам». Дж. Орг. Хим . 67 (4): 1261–1265. дои : 10.1021/jo016096c . ПМИД 11846671 .
- ^ Мартин-Матуте, Б.; Эдин, М.; Богар, К.; Кайнак, ФБ; Беквалль, Ж.-Э. (2005). «Комбинированный катализ рутения (II) и липазы для эффективного динамического кинетического разделения вторичных спиртов. Понимание механизма рацемизации». Дж. Ам. хим. Соц . 127 (64): 8817–8825. дои : 10.1021/ja051576x . ПМИД 15954789 .
- ^ Чой, Ю.К.; Эх, Дж. Х.; Ли, Д.; Лим, ИТ; Юнг, JY; Ким, М.-Ж. (1999). «Динамическое кинетическое разрешение ациклических аллиловых ацетатов с использованием липазы и палладия». Дж. Орг. Хим . 64 (22): 8423–8424. дои : 10.1021/jo990956w . ПМИД 11674772 .
- ^ Ли, Д.; Ха, ЭА; Ким, М.-Дж.; Юнг, ХМ; Кох, Дж. Х.; Парк, Дж. (2000). «Динамическое кинетическое разрешение аллиловых спиртов, опосредованное катализаторами на основе рутения и липазы». Орг. Летт . 2 (15): 2377–2379. дои : 10.1021/ol006159y . ПМИД 10930288 .
- ^ Имс, Дж. (2000). «Параллельные кинетические разрешения». Энджью. хим. Межд. Эд . 39 (5): 885–888. doi : 10.1002/(SICI)1521-3773(20000303)39:5<885::AID-ANIE885>3.0.CO;2-2 . ПМИД 10760881 .
- ^ Дехил, младший; Готор, В. (2002). «Параллельное кинетическое разделение рацемических смесей: новая стратегия получения энантиочистых соединений?». хим. Соц. Преподобный . 31 (6): 365–370. дои : 10.1039/B205280F . ПМИД 12491751 .
- ^ Ведейс, Э.; Юре, М. (2005). «Эффективность неферментативного кинетического разрешения». Энджью. хим. Межд. Эд . 44 (5): 3974–4001. дои : 10.1002/anie.200460842 . ПМИД 15942973 .
- ^ Куросу, М.; Киши, Ю. (1998). «Новый пример оптического разрешения рацемических кетонов, полученных в результате синтеза батрахотоксина». Дж. Орг. Хим . 63 (18): 6100–6101. дои : 10.1021/jo981416m . ПМИД 11672234 .
- ^ Дехил, младший; Готор, В. (2002). «Получение энантиочистых кетонов и спиртов, содержащих четвертичный стереоцентр, путем параллельного кинетического разделения β-кетонитрилов». Дж. Орг. Хим . 67 (5): 1716–1718. дои : 10.1021/jo011092t . ПМИД 11871913 .
- ^ Танака, К.; Фу, GC (2003). «Параллельное кинетическое разрешение 4-алкиналов, катализируемое Rh (I) / Tol-BINAP: синтез энантиообогащенных циклобутанонов и циклопентенонов». Дж. Ам. хим. Соц . 125 (27): 8078–8079. дои : 10.1021/ja035489l . ПМИД 12837058 .
Дальнейшее чтение
[ редактировать ]- Динамическое кинетическое разрешение. Встреча группы Макмиллана. Джейк Винер Линк
- Динамическое кинетическое разрешение: мощный подход к асимметричному синтезу. Эрик Алексанян Встреча супергруппы 30 марта 2005 г. Ссылка
- Динамическое кинетическое разрешение: практическое применение в синтезе. Семинар Валери Келлер, 3-й курс, 1 ноября 2001 г. Ссылка
- Кинетическое разрешение. Литературный семинар группы Дэвида Эбнера Штольца. от 4 июня 2003 г. ссылка
- Кинетическое разрешение. Юго-западная презентация ЮТ. связь