Продажа болезнь
| Продажа болезнь | |
|---|---|
| Другие имена | Расширение B-клеток с помощью NF-KB и T-клеточной анологической болезни |
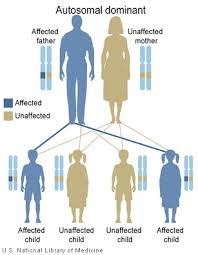 | |
| Для каждого ребенка, которого есть пострадавший родитель, существует 50% вероятность передачи мутации, независимо от пола доминантного доминанта ребенка. | |
| Специальность | Иммунология |
Болезнь Бента является редким генетическим заболеванием иммунной системы . Benta означает « B-клеток расширение с помощью NF-κB и T-клеточной анергии » и вызвано гетерозиготных усилий функции мутациями в генной карте11 ( см. Вход OMIM #607210 ). Это расстройство характеризуется поликлональным В -клеточным лимфоцитозом с началом младенчества, спленомегалией , лимфаденопатией , легким иммунодефицитом и повышенным риском лимфомы . Следователи Эндрю Л. Сноу и Майкл Дж. Ленардо в Национальном институте аллергии и инфекционных заболеваний в Национальном институте здравоохранения США впервые охарактеризовали болезнь Бента в 2012 году. Нынешняя лаборатория доктора Сноу в Университете медицинских наук в форме службы в настоящее время активно активно является активной изучая это расстройство. [ 1 ] [ 2 ]
Презентация
[ редактировать ]Люди с болезнью Бента имеют поликлональный В -клеточный лимфоцитоз (т.е. избыток В -клеток), развивающихся в младенчестве, в дополнение к спленомегалии и лимфаденопатии . Пациенты могут иметь низкую сывороточную IgM и легкие аноргические Т -клетки. Эти особенности, вероятно, способствуют умеренному иммунодефициту , наблюдаемому при болезни Бента. Пациенты, как правило, восприимчивы к рецидивирующим синопульмональным и ушным инфекциям в детстве и могут быть более восприимчивы к определенным вирусам, включая вирус Эпштейна -Барра , вирус BK и контагиоз молтускума . [ 1 ]
Генетика
[ редактировать ]
Болезнь Бента вызвана зародышевой линией мутаций усиления функции в генной карте11 . Это картирование гена 138 КБ с хромосомой 7P22 с 26 экзонами , кодирующими 1154 аминокислотного белка. [ 3 ] [ 4 ] Белок Card11 (также известный как Carma1) представляет собой белок каркасов, необходимый для активации NF-κB как в B, так и в T-лимфоцитах. Мутации усиления функции стимулируют конститутивную активацию NF-κB в обоих типах клеток. Большинство мутаций локализованы в пределах или чуть выше по течению от домена из спиральной камеры (экзоны 4–9) белка. Фенотипы пациентов также предполагают, что дифференцировка В-клеток может быть частично нарушена при заболевании бента, что способствует низкому проценту переключенных классов и В-клеток памяти. [ 1 ] [ 2 ]
Мутации усиления функции зародышевой линии в CARD11 проявляют менее тяжелую болезнь, чем мутации потери функции, наблюдаемая при дефиците карты11 ( OMIM #615206 ), аутосомно-рецессивное состояние , проявляющее в тяжелом комбинированном иммунодефиците . [ 1 ]
Мутации карты увеличения функции11, связанные с болезнью Бента, также могут предрасполагать пациентов к злокачественной опухоле B-клеток . Важно отметить, что гиперактивная NF-κB часто ассоциируется со злокачественной опухолью В-клеток , и, в частности, мутации соматического усиления функции11 часто наблюдаются при диффузной большой В-клеточной лимфоме ( DLBCL ). Тем не менее, большинство пациентов с бентой имеют накопление поликлональных В -клеток без признаков олигоклональных или моноклональных популяций (то есть злокачественная опухоль). Эти мутации, по -видимому, не связаны с злокачественными новообразованиями Т -клеток . [ 2 ]
Наследование
[ редактировать ]Это расстройство унаследовано аутосомно -доминантным образом. Аутосомный относится к тому факту, что у каждого человека есть два аллеля карты , один унаследованный от каждого родителя. Это в отличие от пола, связанных с полом хромосом . Доминирующее означает, что аномальный аллель доминирует в соответствующем, нормальном аллеле. Только одна из двух копий (аллелей) карты11 должна быть ненормальной, чтобы человек имел болезнь бента. Болезнь Бента также может возникнуть спонтанно у пациента в результате de novo мутаций в карте11, что означает, что мутация не была унаследована от родителей. В этом случае пациент все еще мог передать мутация своим детям. [ 5 ]
Дети родителя, который несет мутацию карты11, имеют 50% шанс наследовать мутацию. В семье риск унаследовать аллель Muttited Card11 не зависит от того, унаследовал ли другие братья и сестры унаследовать мутация. Например, если у первых четырех детей в семье есть мутация, у следующего ребенка такой же 50% риск наследства мутации. У детей, которые не наследуют мутацию, не развиваются болезнь Бента и не передадут ее своим детям. [ 5 ]
Диагноз
[ редактировать ]Большинство мононуклетов для периферической крови пациента являются поликлональные наивные зрелые В -клетки со значительным увеличением числа незрелых, переходных В -клеток (идентифицированные как CD10+). [ 6 ] Процент циркулирующих классовых переключателей и В-клеток памяти очень низкие, а исследования in vitro показывают плохую дифференцировку В-клеток и секрецию иммуноглобулина. Сывороточный IGM низкий у большинства пациентов, в то время как общий IgG и IgA могут находиться на нижней части нормального. Пациенты демонстрируют дефектную выработку антител против независимых от Т-клеток вакцин на основе полисахаридов . Некоторые пациенты не могут устанавливать титры защитных антител к другим вакцинам, таким как корь и вирус варицеллы . [ 2 ] [ 7 ]
Количество Т -клеток, как правило, внутри или чуть выше нормального диапазона. in vitro Стимуляция Т -клеток демонстрирует, что как CD4+, так и CD8+ T -клетки менее чувствительны, чем нормы, что предполагает легкую анергию Т -клеток у пациентов. [ 1 ]
Диагноз лейкемии , как правило, можно исключить у этих пациентов на основе ничем не примечательного появления мелких покоящихся лимфоцитов в крови; Тем не менее, пациенты должны быть внимательно следить за любыми признаками экспансии моноклональных или олигоклональных В -клеток, потому что может быть повышенный риск злокачественности В -клеток. В частности, один пациент с болезнью Бента, как сообщалось, развился хронический лимфоцитарный лейкоз (B-CLL) В-клеток (B-CLL) как взрослый. [ 1 ]
Уход
[ редактировать ]There is currently minimal therapeutic intervention available for BENTA disease. Patients are closely monitored for infections and for signs of monoclonal or oligoclonal B cell expansion that could indicate B cell malignancy. Splenectomy is unlikely to reduce B cell burden; peripheral blood B cell counts rose significantly in three patients who underwent the procedure. It remains to be determined whether immunosuppressive drugs, including B cell-depleting drugs such as rituximab, could be effective for treating BENTA disease.[1]
References
[edit]- ^ Jump up to: a b c d e f g Turvey, SE; Durandy, A; Fischer, A; Fung, SY; Geha, RS; Gewies, A; Giese, T; Greil, J; Keller, B; McKinnon, ML; Neven, B; Rozmus, J; Ruland, J; Snow, AL; Stepensky, P; Warnatz, K (2014). "The CARD11-BCL10-MALT1 (CBM) signalosome complex: Stepping into the limelight of human primary immunodeficiency". The Journal of Allergy and Clinical Immunology. 134 (2): 276–84. doi:10.1016/j.jaci.2014.06.015. PMC 4167767. PMID 25087226.
- ^ Jump up to: a b c d e Snow, A. L.; Xiao, W.; Stinson, J. R.; Lu, W.; Chaigne-Delalande, B.; Zheng, L.; Pittaluga, S.; Matthews, H. F.; Schmitz, R.; Jhavar, S.; Kuchen, S.; Kardava, L.; Wang, W.; Lamborn, I. T.; Jing, H.; Raffeld, M.; Moir, S.; Fleisher, T. A.; Staudt, L. M.; Su, H. C.; Lenardo, M. J. (5 November 2012). "Congenital B cell lymphocytosis explained by novel germline CARD11 mutations". Journal of Experimental Medicine. 209 (12): 2247–2261. doi:10.1084/jem.20120831. PMC 3501355. PMID 23129749.
- ^ "CARD11 caspase recruitment domain family, member 11 [ Homo sapiens (human) ]". NCBI > Genes & Expression > Gene. NCBI. Archived from the original on 6 November 2014. Retrieved 4 September 2014.
- ^ "Caspase recruitment domain-containing protein 11 [Homo sapiens]". NCBI. Archived from the original on 6 November 2014. Retrieved 4 September 2014.
- ^ Jump up to: a b "BENTA Disease" (PDF). NIAID: National Institute of Allergy and Infectious Diseases. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES. July 2015. pp. 1–5. Archived (PDF) from the original on 26 March 2023. Retrieved 19 June 2024.
- ^ Chung JB, Silverman M, Monroe JG (June 2003). "Transitional B cells: step by step towards immune competence". Trends Immunol. 24 (6): 343–9. doi:10.1016/s1471-4906(03)00119-4. PMID 12810111.
- ^ Kniffin, Cassandra. "#606445 Persistent polyclonal B-cell lymphocytosis; PPBL". OMIM. Johns Hopkins University. Archived from the original on 24 September 2015. Retrieved 4 September 2014.