Биопитон
| Оригинальный автор(ы) | Чепмен Б., Чанг Дж. [ 1 ] |
|---|---|
| Первоначальный выпуск | 17 декабря 2002 г |
| Стабильная версия | 1.81 [ 2 ] |
| Репозиторий | https://github.com/biopython/biopython [ 3 ] |
| Написано в | Питон и Си |
| Платформа | Кроссплатформенность |
| Тип | Биоинформатика |
| Лицензия | Лицензия Биопитона |
| Веб-сайт | биопитон |
Проект Biopython — это с открытым исходным кодом коллекция некоммерческих инструментов Python для вычислительной биологии и биоинформатики , созданная международной ассоциацией разработчиков. [ 1 ] [ 4 ] [ 5 ] Он содержит классы для представления биологических последовательностей и аннотаций последовательностей , а также способен читать и записывать файлы различных форматов. Это также позволяет использовать программные средства доступа к онлайновым базам данных биологической информации , например, к базам данных NCBI . Отдельные модули расширяют возможности Biopython по выравниванию последовательностей , структуре белков , популяционной генетике , филогенетике , мотивам последовательностей и машинному обучению . Biopython — один из нескольких проектов Bio*, призванных уменьшить дублирование кода в вычислительной биологии . [ 6 ]
История
[ редактировать ]Разработка Biopython началась в 1999 году и впервые была выпущена в июле 2000 года. [ 7 ] Он был разработан в те же сроки и с теми же целями, что и другие проекты, которые добавляли возможности биоинформатики в соответствующие языки программирования, включая BioPerl , BioRuby и BioJava . Среди первых разработчиков проекта были Джефф Чанг, Эндрю Далк и Брэд Чепмен, хотя на сегодняшний день свой вклад внесли более 100 человек. [ 8 ] В 2007 году был создан аналогичный проект Python , а именно PyCogent . [ 9 ]
Первоначальная область применения Biopython включала доступ, индексирование и обработку файлов биологических последовательностей. Хотя этому по-прежнему уделяется большое внимание, в последующие годы добавленные модули расширили свою функциональность, чтобы охватить дополнительные области биологии (см. Основные функции и примеры ).
Начиная с версии 1.77, Biopython больше не поддерживает Python 2. [ 10 ]
Дизайн
[ редактировать ]Везде, где это возможно, Biopython следует соглашениям, используемым в языке программирования Python, чтобы облегчить работу пользователям, знакомым с Python. Например, Seq и SeqRecord объектами можно манипулировать с помощью Slicing , аналогично строкам и спискам Python. Он также функционально аналогичен другим проектам Bio*, таким как BioPerl. [ 7 ]
Biopython может читать и записывать файлы наиболее распространенных форматов для каждой из своих функциональных областей, а его лицензия является разрешительной и совместима с большинством других лицензий на программное обеспечение, что позволяет использовать Biopython в различных программных проектах. [ 5 ]
Ключевые особенности и примеры
[ редактировать ]Последовательности
[ редактировать ]Основной концепцией Biopython является биологическая последовательность, и она представлена Seq сорт. [ 11 ] Биопитон Seq Объект во многих отношениях похож на строку Python: он поддерживает нотацию срезов Python, может быть объединен с другими последовательностями и является неизменяемым. Кроме того, он включает методы, специфичные для последовательностей, и определяет конкретный используемый биологический алфавит.
>>> # This script creates a DNA sequence and performs some typical manipulations
>>> from Bio.Seq import Seq
>>> dna_sequence = Seq("AGGCTTCTCGTA", IUPAC.unambiguous_dna)
>>> dna_sequence
Seq('AGGCTTCTCGTA', IUPACUnambiguousDNA())
>>> dna_sequence[2:7]
Seq('GCTTC', IUPACUnambiguousDNA())
>>> dna_sequence.reverse_complement()
Seq('TACGAGAAGCCT', IUPACUnambiguousDNA())
>>> rna_sequence = dna_sequence.transcribe()
>>> rna_sequence
Seq('AGGCUUCUCGUA', IUPACUnambiguousRNA())
>>> rna_sequence.translate()
Seq('RLLV', IUPACProtein())
Аннотация последовательности
[ редактировать ]The SeqRecord Класс описывает последовательности вместе с такой информацией, как имя, описание и функции, в форме SeqFeature объекты. Каждый SeqFeature Объект определяет тип объекта и его местоположение. Типами объектов могут быть «ген», «CDS» (кодирующая последовательность), «repeat_region», «mobile_element» и другие, а положение объектов в последовательности может быть точным или приблизительным.
>>> # This script loads an annotated sequence from file and views some of its contents.
>>> from Bio import SeqIO
>>> seq_record = SeqIO.read("pTC2.gb", "genbank")
>>> seq_record.name
'NC_019375'
>>> seq_record.description
'Providencia stuartii plasmid pTC2, complete sequence.'
>>> seq_record.features[14]
SeqFeature(FeatureLocation(ExactPosition(4516), ExactPosition(5336), strand=1), type='mobile_element')
>>> seq_record.seq
Seq("GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC...GCC", IUPACAmbiguousDNA())
Ввод и вывод
[ редактировать ]Biopython может читать и записывать в ряд распространенных форматов последовательностей, включая FASTA , FASTQ , GenBank, Clustal, PHYLIP и NEXUS . При чтении файлов описательная информация в файле используется для заполнения членов классов Biopython, например: SeqRecord. Это позволяет конвертировать записи одного формата файла в другие.
Очень большие файлы последовательностей могут превышать ресурсы памяти компьютера, поэтому Biopython предоставляет различные варианты доступа к записям в больших файлах. Их можно полностью загрузить в память в структурах данных Python, таких как списки или словари , обеспечивая быстрый доступ за счет использования памяти. В качестве альтернативы файлы можно считывать с диска по мере необходимости, что снижает производительность, но требует меньше памяти.
>>> # This script loads a file containing multiple sequences and saves each one in a different format.
>>> from Bio import SeqIO
>>> genomes = SeqIO.parse("salmonella.gb", "genbank")
>>> for genome in genomes:
... SeqIO.write(genome, genome.id + ".fasta", "fasta")
Доступ к онлайн-базам данных
[ редактировать ]Через модуль Bio.Entrez пользователи Biopython могут загружать биологические данные из баз данных NCBI. Каждая из функций поисковой системы Entrez доступна через функции этого модуля, включая поиск и загрузку записей.
>>> # This script downloads genomes from the NCBI Nucleotide database and saves them in a FASTA file.
>>> from Bio import Entrez
>>> from Bio import SeqIO
>>> output_file = open("all_records.fasta", "w")
>>> Entrez.email = "[email protected]"
>>> records_to_download = ["FO834906.1", "FO203501.1"]
>>> for record_id in records_to_download:
... handle = Entrez.efetch(db="nucleotide", id=record_id, rettype="gb")
... seqRecord = SeqIO.read(handle, format="gb")
... handle.close()
... output_file.write(seqRecord.format("fasta"))
Филогения
[ редактировать ]
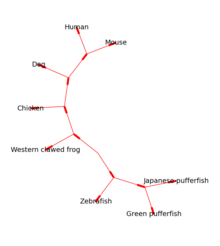
Модуль Bio.Phylo предоставляет инструменты для работы и визуализации филогенетических деревьев . Для чтения и записи поддерживаются различные форматы файлов, включая Newick , NEXUS и phyloXML . Общие манипуляции с деревьями и обходы поддерживаются через Tree и Clade объекты. Примеры включают преобразование и сопоставление файлов дерева, извлечение подмножеств из дерева, изменение корня дерева и анализ таких характеристик ветвей, как длина или оценка. [ 13 ]
Корневые деревья можно рисовать в ASCII или с помощью matplotlib (см. рис. 1), а библиотеку Graphviz можно использовать для создания некорневых макетов (см. рис. 2).
Диаграммы генома
[ редактировать ]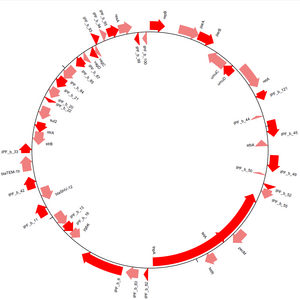
Модуль GenomeDiagram предоставляет методы визуализации последовательностей в Biopython. [ 15 ] Последовательности можно рисовать в линейной или круговой форме (см. рис. 3), поддерживаются многие выходные форматы, включая PDF и PNG . Диаграммы создаются путем создания дорожек и последующего добавления к этим дорожкам функций последовательности. Перебирая элементы последовательности и используя их атрибуты, чтобы решить, будут ли они добавлены к дорожкам диаграммы и каким образом, можно в значительной степени контролировать внешний вид окончательной диаграммы. Между различными треками можно провести перекрестные связи, что позволяет сравнивать несколько последовательностей на одной диаграмме.
Макромолекулярная структура
[ редактировать ]Модуль Bio.PDB может загружать молекулярные структуры из файлов PDB и mmCIF и был добавлен в Biopython в 2003 году. [ 16 ] Structure Объект занимает центральное место в этом модуле и организует структуру макромолекул в иерархическом порядке: Structure объекты содержат Model объекты, содержащие Chain объекты, содержащие Residue объекты, содержащие Atom объекты. Неупорядоченные остатки и атомы получают свои классы. DisorderedResidue и DisorderedAtom, которые описывают их неопределенные позиции.
Используя Bio.PDB, можно перемещаться по отдельным компонентам файла структуры макромолекул, например, исследовать каждый атом белка. Можно проводить общие анализы, такие как измерение расстояний или углов, сравнение остатков и расчет глубины остатков.
Популяционная генетика
[ редактировать ]Модуль Bio.PopGen добавляет поддержку Biopython для Genepop, пакета программного обеспечения для статистического анализа популяционной генетики. [ 17 ] Это позволяет анализировать равновесие Харди-Вайнберга , неравновесие по сцеплению в популяции и другие особенности частот аллелей .
Этот модуль также может выполнять популяционно-генетическое моделирование, используя теорию объединения с программой fastsimcoal2. [ 18 ]
Обертки для инструментов командной строки
[ редактировать ]Многие модули Biopython содержат оболочки командной строки для часто используемых инструментов, что позволяет использовать эти инструменты изнутри Biopython. К этим оболочкам относятся BLAST , Clustal , PhyML, EMBOSS и SAMtools . Пользователи могут создать подкласс общего класса-оболочки, чтобы добавить поддержку любого другого инструмента командной строки.
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б Чепмен, Брэд; Чанг, Джефф (август 2000 г.). «Биопитон: инструменты Python для вычислительной биологии» . Информационный бюллетень ACM SIGBIO . 20 (2): 15–19. дои : 10.1145/360262.360268 . S2CID 9417766 .
- ^ «Выпуск biopython-181: зафиксировать выпуск 1.81 (# 4233)» . Проверено 22 апреля 2023 г.
- ^ Ошибка: невозможно правильно отобразить ссылку. смотрите в документации . Подробности
- ^ Кок, Питер Дж.А.; Антао, Тьяго; Чанг, Джеффри Т; Чепмен, Брэд А; Кокс, Саймон Дж; Далк, Эндрю; Фридберг, Иддо; Хамелрик, Томас; Кауфф, Фрэнк; Вильчинский, Бартек; де Хун, Мишель Дж.Л. (20 марта 2009 г.). «Биопитон: свободно доступные инструменты Python для вычислительной молекулярной биологии и биоинформатики» . Биоинформатика . 25 (11): 1422–3. doi : 10.1093/биоинформатика/btp163 . ПМЦ 2682512 . ПМИД 19304878 .
- ^ Jump up to: а б На веб-сайте Biopython можно найти другие статьи, описывающие Biopython , а также список из более чем ста публикаций, использующих или цитирующих Biopython .
- ^ Мангалам, Гарри (сентябрь 2002 г.). «Наборы инструментов Bio* — краткий обзор» . Брифинги по биоинформатике . 3 (3): 296–302. дои : 10.1093/нагрудник/3.3.296 . ПМИД 12230038 .
- ^ Jump up to: а б Чепмен, Брэд (11 марта 2004 г.), Проект Biopython: Философия, функциональность и факты (PDF) , получено 11 сентября 2014 г.
- ^ Список участников Biopython , заархивировано из оригинала 11 сентября 2014 г. , получено 11 сентября 2014 г.
- ^ Найт, Р; Максвелл, П; Бирмингем, А; Карнс, Дж; Капорасо, Дж.Г.; Истон, Британская Колумбия; Итон, М; Хамади, М; Линдси, Х; Лю, З; Лозупон, С; Макдональд, Д; Робсон, М; Саммут, Р; Смит, С; Уэйкфилд, MJ; Видманн, Дж; Викман, С; Уилсон, С; Ин, Х; Хаттли, Джорджия (2007). «Py Cogent : набор инструментов для понимания последовательности» . Геномная биология . 8 (8): 171 р. дои : 10.1186/gb-2007-8-8-r171 . ПМК 2375001 . ПМИД 17708774 .
- ^ Дэйли, Крис, выпущен Biopython 1.77 , получено 6 октября 2021 г.
- ^ Чанг, Джефф; Чепмен, Брэд; Фридберг, Иддо; Хамелрик, Томас; де Хун, Мишель; Кок, Питер; Антао, Тьяго; Талевич, Эрик; Вильчинский, Бартек (29 мая 2014 г.), Учебное пособие и кулинарная книга по Biopython , получено 28 августа 2014 г.
- ^ Змасек, Кристиан М; Чжан, Цин; Йе, Южен; Годзик, Адам (24 октября 2007 г.). «Удивительная сложность наследственной сети апоптоза» . Геномная биология . 8 (10): 226 р. дои : 10.1186/gb-2007-8-10-r226 . ПМК 2246300 . ПМИД 17958905 .
- ^ Талевич, Эрик; Инверго, Брэндон М; Кок, Питер Дж.А.; Чепмен, Брэд А. (21 августа 2012 г.). «Bio.Phylo: унифицированный набор инструментов для обработки, анализа и визуализации филогенетических деревьев в Biopython» . БМК Биоинформатика . 13 (209): 209. дои : 10.1186/1471-2105-13-209 . ПМЦ 3468381 . ПМИД 22909249 .
- ^ «Плазмида pKPS77 штамма Klebsiella pneumoniae KPS77, полная последовательность» . НКБИ . Проверено 10 сентября 2014 г.
- ^ Причард, Лейтон; Уайт, Дженнифер А; Берч, Пол Р.Дж.; Тот, Ян К. (март 2006 г.). «GenomeDiagram: пакет Python для визуализации крупномасштабных геномных данных» . Биоинформатика . 22 (5): 616–617. doi : 10.1093/биоинформатика/btk021 . ПМИД 16377612 .
- ^ Хамелрик, Томас; Мандерик, Бернар (10 мая 2003 г.). «Парсер файлов PDB и класс структуры, реализованные на Python» . Биоинформатика . 19 (17): 2308–2310. doi : 10.1093/биоинформатика/btg299 . ПМИД 14630660 .
- ^ Руссе, Франсуа (январь 2008 г.). «GENEPOP'007: полная повторная реализация программного обеспечения GENEPOP для Windows и Linux». Ресурсы молекулярной экологии . 8 (1): 103–106. дои : 10.1111/j.1471-8286.2007.01931.x . ПМИД 21585727 . S2CID 25776992 .
- ^ Экскофье, Лоран; Фолль, Матье (1 марта 2011 г.). «fastsimcoal: непрерывный коалесцентный симулятор геномного разнообразия в условиях произвольно сложных эволюционных сценариев» . Биоинформатика . 27 (9): 1332–1334. doi : 10.1093/биоинформатика/btr124 . ПМИД 21398675 .