Синдром Герстмана-Штраусслера-Шейнкера
| Синдром Герстмана-Штраусслера-Шейнкера | |
|---|---|
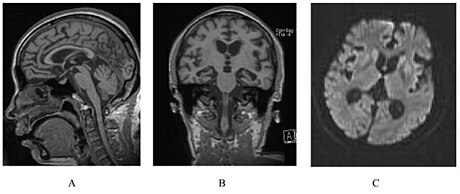 | |
| У человека с наследственной прионной болезнью наблюдается атрофия мозжечка. Это вполне типично для ГСС. | |
| Специальность | Неврология |
| Симптомы | трудности с речью, развитие слабоумия , потеря памяти , потеря зрения . |
| Причины | Прионы |
| Прогноз | Обычно летальный исход, ожидаемая продолжительность жизни обычно составляет 5-6 лет с момента постановки диагноза. |
Синдром Герстмана-Штраусслера-Шейнкера ( СГШ ) — крайне редкое, всегда смертельное (вследствие того, что оно вызывается прионами ) нейродегенеративное заболевание , поражающее пациентов в возрасте от 20 до 60 лет. Он передается исключительно по наследству и встречается лишь в нескольких семьях во всем мире. [ 1 ] Однако его классифицируют как трансмиссивные губчатые энцефалопатии (TSE) из-за причинной роли PRNP , человеческого прионного белка. [ 2 ] Впервые GSS был описан австрийскими врачами Йозефом Герстманном , Эрнстом Штраусслером и Ильей Шейнкером в 1936 году. [ 3 ] [ 4 ]
Семейные случаи связаны с аутосомно -доминантным наследованием. [ 5 ]
Некоторые симптомы являются общими для GSS, например прогрессирующая атаксия , пирамидные знаки и деменция ; они ухудшаются по мере прогрессирования заболевания. [ 6 ]
Симптомы и признаки
[ редактировать ]Симптомы начинаются с медленно развивающейся дизартрии (трудности с речью) и атаксии туловища мозжечка (неустойчивости), а затем прогрессирующая деменция становится более очевидной. На ранних стадиях GSS люди с этим заболеванием также могут проявлять неуклюжесть и испытывать трудности при ходьбе. По мере прогрессирования заболевания симптомы атаксии становятся более выраженными. [ 7 ] Потеря памяти может быть первым симптомом СГС. [ 8 ] заболевание может имитировать спиноцеребеллярную атаксию Могут возникать экстрапирамидные и пирамидные симптомы и признаки, а на начальных стадиях . Миоклонус (спазматическое сокращение мышц) наблюдается реже, чем при болезни Крейтцфельдта-Якоба . У многих пациентов также наблюдается нистагм (непроизвольное движение глаз), нарушения зрения и даже слепота или глухота. [ 9 ] Нейропатологические проявления GSS включают широко распространенное отложение амилоидных бляшек, состоящих из аномально свернутого прионного белка. [ 8 ]
Выделяют четыре клинических фенотипа: типичный GSS, GSS с арефлексией и парестезией, GSS чистой деменции и GSS, подобный болезни Крейтцфельдта-Якоба. [ 10 ]
Причины
[ редактировать ]СГС входит в группу заболеваний, называемых трансмиссивными губчатыми энцефалопатиями . Эти заболевания вызываются прионами — классом патогенных белков, устойчивых к протеазам . Эти прионы затем образуют кластеры в мозге, которые отвечают за нейродегенеративные эффекты, наблюдаемые у пациентов. [ 11 ]
P102L Мутация , которая вызывает замену пролина на лейцин в кодоне 102, была обнаружена в гене прионного белка ( PRNP , на хромосоме 20 ) у большинства больных людей. [ 12 ] Таким образом, похоже, что это генетическое изменение обычно необходимо для развития заболевания. [ 13 ] [ 14 ] [ 15 ]
Диагностика
[ редактировать ]GSS можно выявить с помощью генетического тестирования . [ 9 ] Тестирование на GSS включает исследование крови и ДНК, чтобы попытаться обнаружить мутировавший ген в определенных кодонах. Если присутствует генетическая мутация, у пациента в конечном итоге разовьется GSS.
Уход
[ редактировать ]Лекарства от GSS не существует, как и не существует какого-либо известного лечения, способного замедлить прогрессирование заболевания. Терапия и лекарства направлены на лечение или замедление последствий симптомов. Целью этих методов лечения является попытка максимально улучшить качество жизни пациента. В настоящее время продолжаются исследования по поиску лекарства, одним из наиболее ярких примеров которых является моноклональное антитело PRN100. [ нужна ссылка ]
Прогноз
[ редактировать ]СГС — это заболевание, которое прогрессирует медленно и длится примерно 2–10 лет, в среднем около пяти лет. [ 8 ] [ 1 ]
Симптомы в виде неуклюжести и неустойчивости при ходьбе в начале заболевания. Подергивания мышц (миоклонус) встречаются гораздо реже, чем при болезни Крейтцфельдта-Якоба. Речь становится затрудненной (так называемая дизартрия), развивается деменция. Могут развиться нистагм (быстрое движение глаз в одном направлении с последующим более медленным возвратом в исходное положение) и глухота. Нарушается координация мышц (таксия). Мышцы могут стать жесткими. Обычно повреждаются мышцы, контролирующие дыхание и кашель, что приводит к высокому риску развития пневмонии, которая является наиболее распространенной причиной смерти.
Заболевание в конечном итоге приводит к смерти, чаще всего из-за того, что пациент впадает в кому, или от вторичной инфекции из-за потери пациентом функций организма. [ 1 ]
Исследовать
[ редактировать ]Прионные заболевания, также называемые трансмиссивными губчатыми энцефалопатиями (ТГЭ), представляют собой нейродегенеративные заболевания головного мозга, которые, как считается, вызываются белком, который превращается в аномальную форму, называемую прионом. [ 16 ] [ 17 ] GSS является очень редким TSE, поэтому его генетическое происхождение практически невозможно определить. Также сложно найти пациентов с СГШ, поскольку об этом заболевании, как правило, занижают из-за его клинического сходства с другими заболеваниями, и оно обнаружено лишь в нескольких странах. [ 18 ] В 1989 году в семействе GSS была выявлена первая мутация гена прионного белка. [ 15 ] Самой крупной из этих семей, пострадавших от GSS, является Индиана Киндред, охватывающая более 8 поколений и насчитывающая более 3000 человек, из которых, как известно, пострадало 57 человек. [ 14 ] Позже выяснилось, что GSS имеет множество различных типов генных мутаций, различающихся по тяжести симптомов, времени и прогрессированию. Врачи в разных частях мира находятся в процессе выявления новых поколений и семей, у которых есть мутация. [ нужна ссылка ]
Примечания
[ редактировать ]- ^ Перейти обратно: а б с «Информационная страница о болезни Герстмана-Штрауслера-Шейнкера» . Национальный институт неврологических расстройств и инсульта . Министерство здравоохранения и социальных служб США . Проверено 18 апреля 2021 г.
- ^ Либерский П.П. (2012). «Болезнь Герстмана-Штраусслера-Шейнкера». Нейродегенеративные заболевания . Достижения экспериментальной медицины и биологии. Том. 724. стр. 128–37. дои : 10.1007/978-1-4614-0653-2_10 . ISBN 978-1-4614-0652-5 . ПМИД 22411239 .
- ^ Synd/2269 в Who Named It?
- ^ Герстманн Дж, Штраусслер Э, Шейнкер I (1936). «О своеобразном наследственно-семейном заболевании центральной нервной системы. Заодно вклад в вопрос о преждевременном локальном старении». Журнал для всей неврологии и психиатрии . 154 :736-762. дои : 10.1007/bf02865827 . S2CID 86904496 .
- ^ Де Мишель Дж., Поккьяри М., Петрароли Р., Манфреди М., Каневе Дж., Коппола Дж. и др. (август 2003 г.). «Вариабельный фенотип в итальянской семье P102L Герстмана-Штраусслера-Шейнкера» . Канадский журнал неврологических наук . 30 (3): 233–6. дои : 10.1017/S0317167100002651 . ПМИД 12945948 .
- ^ Фарлоу М.Р., Йи Р.Д., Длоуи С.Р., Коннелли П.М., Аззарелли Б., Гетти Б. (ноябрь 1989 г.). «Болезнь Герстмана-Штраусслера-Шейнкера. I. Расширение клинического спектра». Неврология . 39 (11): 1446–52. дои : 10.1212/wnl.39.11.1446 . ПМИД 2812321 . S2CID 23716392 .
- ^ «Болезнь Герстмана-Штрауслера-Шейнкера» . Национальный институт неврологических расстройств и инсульта . Проверено 27 февраля 2023 г.
- ^ Перейти обратно: а б с Коллинз С., Маклин Калифорния, Мастерс CL (сентябрь 2001 г.). «Синдром Герстмана-Штраусслера-Шейнкера, фатальная семейная бессонница и куру: обзор этих менее распространенных трансмиссивных губчатых энцефалопатий человека». Журнал клинической неврологии . 8 (5): 387–97. дои : 10.1054/jocn.2001.0919 . ПМИД 11535002 . S2CID 31976428 .
- ^ Перейти обратно: а б Гамбетти П. «Болезнь Герстмана-Штраусслера-Шейнкера» . Руководства Merck: Медицинская онлайн-библиотека. Архивировано из оригинала 22 февраля 2011 года . Проверено 6 апреля 2011 г.
- ^ Тесар А, Матей Р, Кукал Дж, Йоханидесова С, Ректорова И, Выхналек М, Келлер Дж, Элиасова И, Паробкова Е, Сметакова М, Мусова З, Русина Р (ноябрь 2019 г.). «Клиническая изменчивость синдрома P102L Герстмана-Штраусслера-Шейнкера». Анналы неврологии . 86 (5): 643–652. дои : 10.1002/ana.25579 . ПМИД 31397917 . S2CID 199504473 .
- ^ Поджиолини И., Савериони Д., Парчи П. (2013). «Неправильное сворачивание прионного белка, штаммы и нейротоксичность: обновленная информация об исследованиях прионов млекопитающих» . Международный журнал клеточной биологии . 2013 : 910314. doi : 10.1155/2013/910314 . ПМЦ 3884631 . ПМИД 24454379 .
- ^ Арата Х., Такашима Х., Хирано Р., Томимицу Х., Мачигашира К., Изуми К. и др. (июнь 2006 г.). «Ранние клинические признаки и результаты визуализации синдрома Герстмана-Штраусслера-Шейнкера (Pro102Leu)». Неврология . 66 (11): 1672–8. дои : 10.1212/01.wnl.0000218211.85675.18 . ПМИД 16769939 . S2CID 26013402 .
- ^ Умех CC, Калакоти П., Гринберг М.К., Нотари С., Коэн Ю., Гамбетти П. и др. (18 февраля 2016 г.). «Клиникопатологические корреляты носителя мутации PRNP P102L с быстро прогрессирующим паркинсонизмом-дистонией» . Клиническая практика двигательных расстройств . 3 (4): 355–358. дои : 10.1002/mdc3.12307 . ПМК 5015693 . ПМИД 27617269 .
- ^ Перейти обратно: а б Гетти Б., Пиккардо П., Франджионе Б., Буджиани О., Джакконе Дж., Янг К. и др. (апрель 1996 г.). «Прионный белковый амилоидоз» . Патология головного мозга . 6 (2): 127–45. дои : 10.1111/j.1750-3639.1996.tb00796.x . ПМИД 8737929 . S2CID 10240829 .
- ^ Перейти обратно: а б Сяо К., Бейкер Х.Ф., Кроу Т.Дж., Поултер М., Оуэн Ф., Тервиллигер Дж.Д. и др. (март 1989 г.). «Связь миссенс-варианта прионного белка с синдромом Герстмана-Штраусслера». Природа . 338 (6213): 342–5. Бибкод : 1989Natur.338..342H . дои : 10.1038/338342a0 . ПМИД 2564168 . S2CID 4319741 .
- ^ Цзоу WQ, Гамбетти П., Сяо X, Юань Дж., Лангевелд Дж., Пирисину Л. (июль 2013 г.). «Прионы при прионопатии с различной чувствительностью к протеазам: обновленная информация» . Патогены . 2 (3): 457–71. doi : 10.3390/pathogens2030457 . ПМЦ 4235694 . ПМИД 25437202 .
- ^ Гамбетти П. (март 2013 г.). «Креационизм и эволюционизм в прионах» . Американский журнал патологии . 182 (3): 623–7. дои : 10.1016/j.ajpath.2012.12.016 . ПМК 3590995 . ПМИД 23380581 .
- ^ Гетти Б., Тальявини Ф., Такао М., Буджиани О., Пиккардо П. (март 2003 г.). «Наследственные амилоидозы прионного белка». Клиники лабораторной медицины . 23 (1): 65–85, viii. дои : 10.1016/s0272-2712(02)00064-1 . ПМИД 12733425 .
Внешние ссылки
[ редактировать ]- Синдром Герстмана-Штраусслера-Шейнкера. Архивировано 11 мая 2013 г. в Wayback Machine , MedicineNet.com.
- ИТАЛЬЯНСКАЯ АССОЦИАЦИЯ ПРОТИВ БОЛЕЗНИ ГЕРСТМАНА ШТРАУССЛЕРА ШЕЙНКЕРА