α-Галокетон

В органической химии представляет α-галогенкетон собой функциональную группу, состоящую из кетоновой группы или, в более общем смысле, карбонильной группы с α- галогеновым заместителем . α-Галогенкетоны являются алкилирующими агентами . Выдающиеся α-галогенкетоны включают фенацилбромид и хлорацетон . [1]
Структура
[ редактировать ]Общая структура: RR'C(X)C(=O)R, где R представляет собой алкильный или арильный остаток, а X - любой из галогенов. Предпочтительной конформацией галогенкетона является цисоид, в котором галоген и карбонил находятся в одной плоскости, поскольку стерические препятствия с карбонилалкильной группой обычно больше. [2]
Синтез галогенкетонов
[ редактировать ]Галогенкетоны и галогенкарбонильные соединения обычно синтезируются путем реакции карбонильных соединений с источниками X. + (X = галоген), который обеспечивается с помощью галогенов : [1]
- RC(O)CH 3 + X 2 → RC(O)CH 2 X + HX
Специализированные источники электрофильных галогенирующих агентов включают N -бромсукцинимид и 1,3-дибром-5,5-диметилгидантоин (DBDMH). В реакции Ниренштейна ацилхлорид реагирует с диазометаном.
Асимметричный синтез
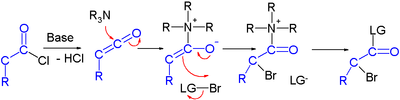
[ редактировать ]Сообщается об усилиях по асимметричному синтезу галогенкарбонилов посредством органокатализа . В одном исследовании хлорангидрид превращается в α-галогеновый эфир с сильным основанием ( гидридом натрия ), донором брома и органокатализатором на основе пролина и хинина : [3]

В предлагаемом механизме реакции основание сначала превращает хлорангидрид в кетен , затем органокатализатор вводит хиральность через свой хиноноидный третичный амин , образуя аддукт кетена.
Реакции
[ редактировать ]Показательны их алкилирующая активность реакции с йодидом калия в ацетоне , хлорацетон реагирует быстрее, чем 1-хлорпропан , в 36 000 раз. Галогенкетоны реагируют с фосфитами по реакции Перкова .
Галогеновую группу можно удалить при восстановительном дегалогенировании галогенкетонов . α-Галогенкетоны также можно превратить в алкены обработкой гидразином.
Благодаря наличию двух электроноакцепторных групп (карбонильной и галогенидной) α-водород является кислым. Это свойство используется в перегруппировке Фаворского , где основание сначала отрывает кислый α-водород, а образующийся карбанион затем замещает галоген.
В перекрестных альдольных реакциях между галогенкетонами и альдегидами первоначальным продуктом реакции является галогенгидрин , который впоследствии может образовывать оксиран в присутствии основания.
α-Галогенкетоны могут реагировать с аминами с образованием α-галогенимина, который можно превратить обратно в исходный галогенкетон путем гидролиза , так что галогенимины можно использовать в качестве замаскированных версий галогенкетонов. Это позволяет осуществить некоторые химические превращения, которые невозможны непосредственно с исходными галогенкетонами. [4]
Предшественники гетероциклов
[ редактировать ]Галогенкетоны принимают участие в нескольких типах реакций, тем более что они бифункциональны, с двумя электрофильными центрами (α-углеродом и карбонильным углеродом). В одном из проявлений этой двойственности они являются предшественниками гетероциклов. Тиазолы возникают в результате реакции хлорацетона с тиоамидами. 2-Аминотиазолы получают аналогичным образом реакцией 2-хлоркетонов с тиомочевиной . [5] [6] Пирролы могут быть синтезированы реакцией галогенкетонов с дикарбонилами и аммиаком в синтезе пиррола Ганча .
Ссылки
[ редактировать ]- ^ Jump up to: а б Вере, Роланд; Де Кимпе, Норберт (1983). «Синтез и реакционная способность α-галогенированных кетонов». У Саула Патая, Цви Раппопорта (ред.). Галогениды, псевдогалогениды и азиды: Vol. 1 . Химия функциональных групп ПАТАИ. стр. 813–931. дои : 10.1002/9780470771716.ch19 . ISBN 9780470771716 .
- ^ Эриан, Айман В.; Шериф, Шериф М.; Габер, Хатем М. (2003). «Химия α-галокетонов и их использование в гетероциклическом синтезе» (PDF) . Молекулы . 8 (11): 793–865. дои : 10.3390/81100793 . S2CID 53951565 .
- ^ Дого-Изонажи, Каэтан; Бекеле, Тефсит; Франция, Стефан; Вулфер, Джеймисон; Уэтервакс, Энтони; Тагги, Эндрю Э.; Лекка, Томас (2006). «Масштабируемая методология каталитического асимметричного α-бромирования хлоридов кислот». Журнал органической химии . 71 (23): 8946–8949. дои : 10.1021/jo061522l . ПМИД 17081026 .
- ^ Вере, Роланд; Де Кимпе, Норберт (1983). «α-галогенированные имины». У Саула Патая, Цви Раппопорта (ред.). Галогениды, псевдогалогениды и азиды: Vol. 1 . Химия функциональных групп ПАТАИ. стр. 813–931. дои : 10.1002/9780470771716.ch13 . ISBN 9780470771716 .
- ^ Дж. Р. Байерс; Дж. Б. Дики (1939). «2-Амино-4-метилтиазол». Органические синтезы . 19:10 . дои : 10.15227/orgsyn.019.0010 .
- ^ Джордж Шварц (1945). «2,4-Диметилтиазол». Органические синтезы . 25:35 . дои : 10.15227/orgsyn.025.0035 .