Органическая фотохимия
Органическая фотохимия включает в себя органические реакции , вызываемые действием света. [1] [2] Поглощение ультрафиолетового света органическими молекулами часто приводит к реакциям. Раньше использовался солнечный свет, а в наше время используются ультрафиолетовые лампы. Органическая фотохимия оказалась очень полезным синтетическим инструментом. Сложные органические продукты можно получить просто.
История
[ редактировать ]Ранние примеры часто обнаруживались путем наблюдения за осадками или изменениями цвета образцов, подвергшихся воздействию солнечного света. Первый случай, о котором сообщил Чамициан, заключался в том, что солнечный свет превращал сантонин в желтый фотопродукт: [3]

Ранним примером осадка была фотодимеризация антрацена , охарактеризованная Юлием Федоровичем Фриче и подтвержденная Эльбсом. [4] Подобные наблюдения были сосредоточены на димеризации коричной кислоты в труксиловую кислоту . В настоящее время известны многие фотодимеры, например димер пиримидина , тиофосген , диамантан .
Другой пример был обнаружен Эгбертом Хавингой в 1956 году. [5] Любопытным результатом стала активация при фотолизе мета-нитрогруппой в отличие от обычной активации орто- и парагруппами.

Органическая фотохимия продвинулась вперед с разработкой правил Вудворда-Гоффмана . [6] [7] Показательно, что эти правила помогают рационализировать фотохимически обусловленное электроциклическое замыкание кольца гекса-2,4-диена, которое происходит дисротационным образом.
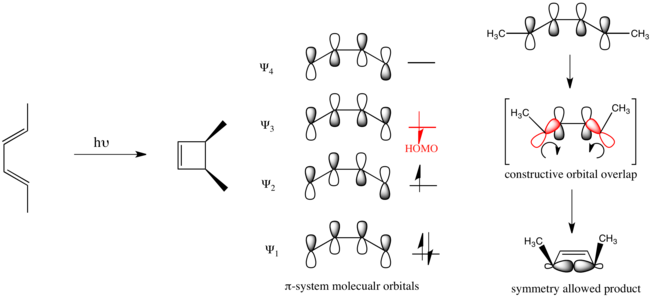
Органические реакции, подчиняющиеся этим правилам, называются разрешенными по симметрии. Реакции, идущие противоположным путем, запрещены по симметрии и требуют значительно больше энергии, чтобы протекать, если они вообще происходят.
Ключевые реакции
[ редактировать ]Органические фотохимические реакции объясняются с точки зрения соответствующих возбужденных состояний . [8] [9]
Параллельно со структурными исследованиями, описанными выше, оценивалась роль спиновой множественности – синглетной или триплетной – на реакционную способность. Подчеркнута важность триплетно-возбужденных видов. Триплеты, как правило, живут дольше, чем синглеты, и имеют более низкую энергию, чем синглет той же конфигурации. Триплеты могут возникать в результате (А) преобразования первоначально образовавшихся синглетов или (Б) взаимодействия с триплетом с более высокой энергией (сенсибилизация).
Возможно гашение триплетных реакций. [10]
Обычные органические фотохимические реакции включают: тип Норриша I , тип Норриша II , рацемизацию оптически активных бифенилов, циклогексадиеноновую перегруппировку типа А, циклогексеноновую перегруппировку типа В, ди -π -метановую перегруппировку , бициклотип В. перегруппировка ]гексанона в фенолы, фотохимические электроциклические процессы, перегруппировка эпоксикетонов в бета-дикетоны, раскрытие цикла циклопропилкетонов, гетеролиз 3,5-диметоксилбензильных производных и фотохимические циклизации диенов.
Практические соображения
[ редактировать ]
Реагенты фотореакций могут быть как газообразными, так и жидкими. [11] В общем, необходимо подносить реагенты близко к источнику света, чтобы получить максимально возможную светоотдачу . Для этой цели реакционную смесь можно облучать либо непосредственно, либо в проточном боковом рукаве реактора подходящим источником света. [12]
Недостатком фотохимических процессов является низкая эффективность преобразования электрической энергии в энергию излучения необходимой длины волны . Помимо излучения, источники света генерируют много тепла, что, в свою очередь, требует энергии охлаждения. Кроме того, большинство источников света излучают полихроматический свет, хотя только монохроматический свет . необходим [13] Однако высокий квантовый выход компенсирует эти недостатки.
Работа при низких температурах выгодна, поскольку позволяет избежать побочных реакций (поскольку повышается селективность) и увеличивается выход (поскольку газообразные реагенты меньше вытесняются из растворителя).
Исходные материалы иногда можно охладить перед реакцией до такой степени, что тепло реакции поглощается без дальнейшего охлаждения смеси. В случае газообразного или низкокипящего исходного сырья необходима работа под избыточным давлением. Из-за большого количества возможного сырья описано большое количество процессов. [14] [15] Крупномасштабные реакции обычно проводят в реакторе с мешалкой , барботажном колонном реакторе или трубчатом реакторе с последующей дальнейшей обработкой в зависимости от целевого продукта. [16] В случае реактора с мешалкой лампа (обычно имеющая форму удлиненного цилиндра) снабжается охлаждающей рубашкой и помещается в реакционный раствор. Трубчатые реакторы изготавливаются из кварцевых или стеклянных трубок, которые облучаются снаружи. Преимущество использования реактора с мешалкой заключается в том, что свет не теряется в окружающую среду. Однако интенсивность света быстро падает с увеличением расстояния до источника света из-за адсорбции реагентов. [12]
Влияние излучения на скорость реакции часто можно представить степенным законом, основанным на плотности квантового потока, т.е. мольного кванта света (ранее измерявшегося в единицах Эйнштейна ) на площадь и время. Поэтому одной из целей проектирования реакторов является определение экономически наиболее выгодных размеров с точки зрения оптимизации плотности квантового тока. [17]
Тематические исследования
[ редактировать ][2+2] Циклодополнения
[ редактировать ]Олефины димеризуются под действием УФ-излучения. [18]
Перегруппировка 4,4-дифенилциклогексадиенона
[ редактировать ]Совершенно параллельно примеру с сантонином в люмисантонин происходит перегруппировка 4,4-дифенилциклогексадиенона. [9] возбужденное состояние n-pi* Здесь триплетное подвергается той же бета-бета-связи. За этим следует межсистемное пересечение (т.е. ISC) с образованием синглетного основного состояния, которое рассматривается как цвиттер-ион . Последним шагом является перегруппировка в бициклический фотопродукт. Реакция называется циклогексадиеноновой перегруппировкой типа А.

4,4-дифенилциклогексенон
[ редактировать ]Чтобы предоставить дополнительные доказательства механизма диенона, в котором существует связь между двумя двойными связями,Здесь представлен случай 4,4-дифенилциклогексенона. Видно, что перегруппировка совершенно иная; таким образом, для перегруппировки типа А необходимы две двойные связи. При наличии одной двойной связи одна из фенильных групп, первоначально находившаяся в положении С-4, мигрировала к С-3 (т.е. к бета-углероду). [19]

Когда одна из арильных групп имеет парациано- или пара-метоксигруппу, эта замещенная арильная группа мигрирует преимущественно. [20] Исследование альтернативных видов типа фенония, в которых арильная группа начала мигрировать к бета-углероду, выявило большую делокализацию электрона с пара-заместителем в мигрирующей арильной группе и, следовательно, более стабилизированный путь.

π - π * реактивность
[ редактировать ]Еще одним типом фотохимической реакции является ди -π -метановая перегруппировка . [21] Еще двумя ранними примерами была перегруппировка 1,1,5,5-тетрафенил-3,3-диметил-1,4-пентадиена (молекула «Мариано»). [22] и перегруппировка баррелена в семибуллвален . [23] Отметим, что в отличие от циклогексадиеноновых реакций, в которых используются n- π *-возбужденные состояния, в ди- π -метановых перегруппировках используются π - π *-возбужденные состояния.

Связанные темы
[ редактировать ]Фотоокислительно-восстановительный катализ
[ редактировать ]При фотоокислительно-восстановительном катализе фотон поглощается сенсибилизатором (молекулой или ионом антенны), который затем вызывает окислительно-восстановительные реакции на органическом субстрате. Распространенным сенсибилизатором является трис(бипиридин) рутения(II) . Иллюстрацией фотоокислительно-восстановительного катализа являются некоторые реакции аминотрифторметилирования. [24]

Фотохлорирование
[ редактировать ]Фотохлорирование — одно из крупнейших применений фотохимии в органическом синтезе. Однако фотон поглощается не органическим соединением, а хлором . Фотолиз Cl 2 дает атомы хлора, которые отрывают атомы H от углеводородов, что приводит к хлорированию.



Ссылки
[ редактировать ]- ^ П. Клан, Дж. Вирц Фотохимия органических соединений: от концепций к практике . Уайли, Чичестер, 2009 г., ISBN 978-1405190886 .
- ^ Н. Дж. Турро, В. Рамамурти, Дж. К. Скайано Современная молекулярная фотохимия органических молекул . Университетские научные книги, Саусалито, 2010 г., ISBN 978-1891389252 .
- ^ Рот, Хайнц Д. (1989). «Начало органической фотохимии». Angewandte Chemie International Edition на английском языке . 28 (9): 1193–1207. дои : 10.1002/anie.198911931 .
- ^ Эльбс, Карл (30 июня 1891 г.). «О парантраценах» . Журнал практической химии . 44 (1): 467–469. дои : 10.1002/prac.18910440140 . ISSN 0021-8383 .
- ^ Хавинга, Э.; Де Йонг, Р.О.; Дорст, В. (1956). «Фотохимическое ускорение гидролиза нитрофенилфосфатов и нитрофенилсульфатов». Recueil des Travaux Chimiques des Pays-Bas . 75 (4): 378–383. дои : 10.1002/recl.19560750403 .
- ^ Вудворд, РБ; Хоффманн, Роальд (1969). «Сохранение орбитальной симметрии». Энджью. хим. Межд. Эд . 8 (11): 781–853. дои : 10.1002/anie.196907811 .
- ^ Вудворд, РБ; Хоффманн, Роальд (1971). Сохранение орбитальной симметрии (3-е издание, 1-е изд.). Вайнхайм, BRD: Verlag Chemie GmbH (BRD) и Academic Press (США). стр. 1–178. ISBN 978-1483256153 .
- ^ «Фотохимическая перегруппировка 4,4-дифенилциклогексадиенона. Статья I по общей теории фотохимических реакций», Циммерман, HE; Шустер, DIJ Am. хим. Сок., 1961, 83, 4486–4487.
- ↑ Перейти обратно: Перейти обратно: а б Циммерман, Ховард Э.; Дэвид И. Шустер (1962). «Новый подход к механистической органической фотохимии. IV. Фотохимические перегруппировки 4,4-дифенилциклогексадиенона». Журнал Американского химического общества . 84 (23). АКС: 4527–4540. дои : 10.1021/ja00882a032 .
- ^ «Теренин А.; Ермолаев В. Сенсибилизированная фосфоресценция в органических растворах при низкой температуре; перенос энергии между триплетными состояниями», Пер. Фарадей Общество, 1956, 52, 1042–1052.
- ^ Марио Скьявелло (Hrsg.): Фотоэлектрохимия, фотокатализ и основы фотореакторов и разработки . Спрингер Нидерланды, 2009 г., ISBN 978-90-481-8414-9 , с. 564.
- ↑ Перейти обратно: Перейти обратно: а б Мартин Фишер: Промышленное применение фотохимического синтеза. В: Angewandte Chemie International Edition на английском языке. 17, 1978, стр. 16–26, doi:10.1002/anie.197800161 .
- ^ Дитер Верле, Михаэль В. Тауш, Вольф-Дитер Шторер: Фотохимия: концепции, методы, эксперименты . Уайли и сыновья, 1998 г., ISBN 978-3-527-29545-6 , стр. 271–275.
- ^ Грант США 1379367 , F. Sparre & WE Masland, «Процесс хлорирования», выдан 24 мая 1921 г., передан компании Du Pont.
- ^ Грант США 1459777 , Р. Лейзер и Ф. Зиффер, «Процесс и устройство для хлорирования метана», выдан 14 февраля 1920 г., передан Зифферу Фрицу и Лейзеру Ричарду.
- ^ Дэвид А. Миксон, Майкл П. Борер, Патрисия А. О'Хара: Ультраочистка SiCl4 путем фотохлорирования в барботажном колонном реакторе. В: Журнал Айше. 36, 1990, стр. 216–226, doi:10.1002/aic.690360207 .
- ^ Х. Хартиг: Простое определение размеров фотохимических реакторов . В: Технология инженера-химика – CIT . 42, 1970, стр. 1241–1245, два : 10.1002/cite.330422002 .
- ^ Каргилл1, РЛ; Далтон-младший; Мортон, штат Джорджия; Колдуэлл1, МЫ (1984). «Фотоциклизация енона в алкен: 6-метилбицикло[4.2.0]октан-2-он». Органические синтезы . 62 : 118. дои : 10.15227/orgsyn.062.0118 .
{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка ) - ^ «Механистическая и исследовательская органическая фотохимия, IX. Миграция фенила при облучении 4,4-дифенилциклогексенона», Циммерман, HE; Уилсон, JWJ Am. хим. Сок., 1964, 86, 4036–4042.
- ^ «Фотохимические миграционные способности циклогексенонов. Механистическая и исследовательская органическая фотохимия. XXIII», Циммерман, HE; Рике, РД; Шеффер, JRJ Am. хим. Сок., 1967, 89, 2033–2047.
- ^ «Несимметричное замещение и направление ди-пи-метановой перегруппировки; Механистическая и исследовательская органическая фотохимия. LVI», Циммерман, HE; Пратт, ACJ Am. хим. Соц., 1970, 92, 6259–6267.
- ^ «Перегруппировка ди-пи-метана. Взаимодействие электронно-возбужденных виниловых хромофоров. Циммерман, HE; Мариано, PSJ Am. Chem. Soc., 1969, 91, 1718–1727.
- ^ Циммерман, HE; Грюневальд, Г.Л. (1966). «Химия баррелена. III. Уникальная фотоизомеризация в семибуллвален». Дж. Ам. хим. Соц. 88 (1): 183–184. два : 10.1021/ja009
- ^ Ясу, Юске; Койке, Такаши; Акита, Мунетака (17 сентября 2012 г.). «Трехкомпонентное окситрифторметилирование алкенов: высокоэффективная и региоселективная дифункционализация связей C=C, опосредованная фотоокислительно-восстановительными катализаторами». Angewandte Chemie, международное издание . 51 (38): 9567–9571. дои : 10.1002/anie.201205071 . ПМИД 22936394 .