Фотоокислительно-восстановительный катализ
Эта статья требует внимания эксперта в области химии . Добавьте в этот шаблон причину или параметр обсуждения , чтобы объяснить проблему со статьей. ( январь 2024 г. ) |
Фотоокислительно-восстановительный катализ — это раздел фотохимии , в котором используется одноэлектронный перенос . Фотоокислительно-восстановительные катализаторы обычно изготавливаются из трех классов материалов: комплексов переходных металлов, органических красителей и полупроводников . Хотя органические фотоокислительно-восстановительные катализаторы доминировали на протяжении 1990-х и начала 2000-х годов, [ 1 ] сегодня чаще используются растворимые комплексы переходных металлов.
Фотохимия сенсибилизаторов переходных металлов
[ редактировать ]Сенсибилизаторы поглощают свет, создавая окислительно-восстановительные возбужденные состояния. Для многих сенсибилизаторов на основе металлов возбуждение реализуется как перенос заряда от металла к лиганду , при котором электрон перемещается от металла (например, ад-орбиталь) к орбитали, локализованной на лигандах (например, π*-орбиталь ароматического лиганда). ). Первоначальное возбужденное электронное состояние релаксирует до синглетного возбужденного состояния с наименьшей энергией посредством внутренней конверсии — процесса, при котором энергия рассеивается в виде колебательной энергии, а не в виде электромагнитного излучения. Это синглетное возбужденное состояние может далее релаксировать за счет двух различных процессов: катализатор может флуоресцировать , излучая фотон и возвращаясь в синглетное основное состояние, или он может перейти в триплетное возбужденное состояние с наименьшей энергией (состояние, в котором два неспаренных электрона имеют одинаковый спин). ) вторым безызлучательным процессом, называемым межкомбинационным пересечением .
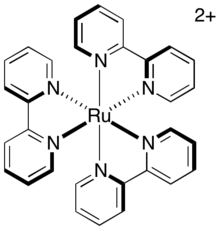
Прямая релаксация возбужденного триплета в основное состояние, называемая фосфоресценцией , требует как испускания фотона, так и инверсии спина возбужденного электрона. Этот путь медленный, поскольку он запрещен по спину , поэтому триплетное возбужденное состояние имеет значительное среднее время жизни. Для обычного фотосенсибилизатора используется трис-(2,2'-бипиридил)рутений (сокращенно [Ru(bipy) 3 ] 2+ или [Ru(bpy) 3 ] 2+ ), время жизни триплетного возбужденного состояния составляет примерно 1100 нс. Этого времени жизни достаточно для того, чтобы другие пути релаксации (в частности, пути переноса электрона) возникли до распада катализатора до его основного состояния.

Долгоживущее триплетное возбужденное состояние, доступное при фотовозбуждении, является одновременно более сильным восстановителем и более сильным окислителем , чем основное состояние катализатора. Поскольку сенсибилизатор является координационно насыщенным, перенос электронов должен происходить посредством процесса внешней сферы , когда электроны туннелируют между катализатором и подложкой.
Перенос электрона во внешнюю сферу
[ редактировать ]Маркуса Теория переноса электронов во внешней сфере предсказывает, что такой туннельный процесс будет происходить наиболее быстро в системах, где перенос электронов термодинамически выгоден (т.е. между сильными восстановителями и окислителями) и где перенос электронов имеет низкий внутренний барьер.
Внутренний барьер переноса электрона вытекает из принципа Франка-Кондона , утверждающего, что электронный переход происходит быстрее при большем перекрытии между начальным и конечным электронными состояниями. В широком смысле этот принцип предполагает, что барьер электронного перехода связан со степенью, в которой система стремится к реорганизации. Для электронного перехода с системой барьер связан с «перекрытием» начальной и конечной волновых функций возбужденного электрона, то есть со степенью, в которой электрон должен «перемещаться» при переходе.
При межмолекулярном переносе электронов аналогичную роль играет степень, в которой ядра стремятся двигаться в ответ на изменение своего нового электронного окружения. Сразу после переноса электрона ядерное расположение молекулы, ранее находившееся в равновесии, теперь представляет собой колебательно-возбужденное состояние и должно релаксировать до своей новой равновесной геометрии. Таким образом, жесткие системы, геометрия которых не сильно зависит от степени окисления, испытывают меньшее колебательное возбуждение во время переноса электрона и имеют более низкий внутренний барьер. Фотокатализаторы, такие как [Ru(bipy) 3 ] 2+ , удерживаются в жестком расположении плоскими бидентатными лигандами, расположенными в октаэдрической геометрии вокруг металлического центра. Следовательно, комплекс не претерпевает значительной реорганизации при переносе электрона. Поскольку перенос электрона в этих комплексах происходит быстро, он, вероятно, будет происходить в течение времени активного состояния катализатора, т.е. в течение времени жизни триплетного возбужденного состояния.

Регенерация катализатора
[ редактировать ]Чтобы регенерировать основное состояние, катализатор должен участвовать во втором внешнесферном переносе электрона. Во многих случаях этот перенос электрона происходит с использованием стехиометрического двухэлектронного восстановителя или окислителя, хотя в некоторых случаях на этом этапе используется второй реагент.
Поскольку стадия переноса электрона каталитического цикла происходит из триплетного возбужденного состояния, она конкурирует с фосфоресценцией как путь релаксации. В экспериментах Штерна-Фольмера измеряется интенсивность фосфоресценции при варьировании концентрации каждого возможного тушащего агента. Изменение концентрации фактического тушащего агента влияет на скорость переноса электронов и степень фосфоресценции. Эта связь моделируется уравнением:
![{\displaystyle \left({\frac {I_{0}}{I}}\right)=1+{k_{q}}*{\tau _{0}}\times [Q]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/338eb04d84052783d691791ccf5c329070594aa0)
Здесь I и I 0 обозначают интенсивность излучения с присутствием и без присутствия гасящего агента, k q - константа скорости процесса тушения, τ 0 - время жизни возбужденного состояния в отсутствие гасящего агента и [Q] концентрация гасящего агента. Таким образом, если время жизни фоторедокс-катализатора в возбужденном состоянии известно из других экспериментов, константу скорости тушения в присутствии одного компонента реакции можно определить, измеряя изменение интенсивности излучения при изменении концентрации гасящего агента.
Фотофизические свойства
[ редактировать ]Редокс-потенциалы
[ редактировать ]Окислительно-восстановительные потенциалы фотоокислительно-восстановительных катализаторов должны быть согласованы с другими компонентами реакции. Хотя окислительно-восстановительные потенциалы основного состояния легко измерить с помощью циклической вольтамперометрии или других электрохимических методов, измерение окислительно-восстановительного потенциала электронно-возбужденного состояния не может быть выполнено непосредственно этими методами. [ 2 ] Однако существуют два метода, которые позволяют оценивать окислительно-восстановительные потенциалы возбужденного состояния, и один метод существует для прямого измерения этих потенциалов. Чтобы оценить окислительно-восстановительные потенциалы возбужденного состояния, один из методов состоит в сравнении скоростей переноса электронов из возбужденного состояния в ряд реагентов в основном состоянии, окислительно-восстановительные потенциалы которых известны. Более распространенный метод оценки этих потенциалов — использовать уравнение, разработанное Ремом и Веллером, которое описывает потенциалы возбужденного состояния как поправку к потенциалам основного состояния:


В этих формулах E* 1/2 представляет собой восстановительный или окислительный потенциал возбужденного состояния, E 1/2 представляет собой восстановительный или окислительный потенциал основного состояния, E 0,0 представляет собой разницу в энергии между нулевыми колебательными состояниями основное и возбужденное состояния, а w r представляет собой работу выхода , электростатическое взаимодействие, возникающее из-за разделения зарядов, происходящего во время переноса электрона между двумя химическими соединениями. Нулевая энергия возбуждения E 0,0 обычно аппроксимируется соответствующим переходом в спектре флуоресценции. Этот метод позволяет рассчитать приблизительные окислительно-восстановительные потенциалы возбужденного состояния на основе более легко измеряемых окислительно-восстановительных потенциалов основного состояния и спектроскопических данных.
Прямое измерение окислительно-восстановительных потенциалов возбужденного состояния возможно с помощью метода, известного как фазомодулированная вольтамперометрия . Этот метод работает, направляя свет на электрохимическую ячейку, чтобы генерировать желаемые виды в возбужденном состоянии, но модулировать интенсивность света синусоидально , так что концентрация частиц в возбужденном состоянии не является постоянной. Фактически, концентрация частиц в возбужденном состоянии в ячейке должна изменяться точно по фазе с интенсивностью света, падающего на электрохимическую ячейку. Если приложенный к клетке потенциал достаточно силен для того, чтобы произошел перенос электрона, изменение концентрации окислительно-восстановительного возбужденного состояния можно измерить как переменный ток (AC). Более того, фазовый сдвиг переменного тока относительно интенсивности падающего света соответствует среднему времени жизни частиц в возбужденном состоянии до того, как они начнут перенос электронов.
Для быстрого доступа доступны диаграммы окислительно-восстановительных потенциалов для наиболее распространенных фотоокислительно-восстановительных катализаторов. [ 3 ]
Электроотрицательность лиганда
[ редактировать ]Относительную восстановительную и окислительную природу этих фотокатализаторов можно понять, приняв во внимание электроотрицательность лигандов и металлоцентр каталитического комплекса. Более электроотрицательные металлы и лиганды могут стабилизировать электроны лучше, чем их менее электроотрицательные аналоги. Следовательно, комплексы с более электроотрицательными лигандами более окисляющие, чем комплексы с менее электроотрицательными лигандами. Например, лиганды 2,2'-бипиридин и 2,2'-фенилпиридин представляют собой изоэлектронные структуры, содержащие одинаковое количество и расположение электронов. Фенилпиридин заменяет один из атомов азота в бипиридине на атом углерода. Углерод менее электроотрицательен, чем азот, поэтому он менее прочно удерживает электроны. Поскольку остальная часть молекулы лиганда идентична, а фенилпиридин удерживает электроны менее прочно, чем бипиридин, он является более сильным электронодонором и менее электроотрицательным как лиганд. Следовательно, комплексы с фенилпиридиновыми лигандами более сильно восстанавливают и менее сильно окисляют, чем эквивалентные комплексы с бипиридиновыми лигандами.
Точно так же фторированный фенилпиридиновый лиганд более электроотрицательен, чем фенилпиридин, поэтому комплексы с фторсодержащими лигандами более сильно окисляют и менее сильно восстанавливают, чем эквивалентные незамещенные фенилпиридиновые комплексы. Электронное влияние металлоцентра на комплекс более сложное, чем лигандное влияние. Согласно Полинга шкале электроотрицательности , и рутений , и иридий имеют электроотрицательность 2,2. Если бы это был единственный фактор, имеющий отношение к окислительно-восстановительным потенциалам, то комплексы рутения и иридия с одними и теми же лигандами должны были бы быть столь же мощными фотоокислительно-восстановительными катализаторами. Однако, учитывая уравнение Рема-Веллера, спектроскопические свойства металла играют роль в определении окислительно-восстановительных свойств возбужденного состояния. [ 4 ] В частности, параметр Е 0,0 связан с длиной волны излучения комплекса и, следовательно, с величиной стоксова сдвига - разницы в энергии между максимальным поглощением и излучением молекулы. Обычно комплексы рутения имеют большие стоксовы сдвиги и, следовательно, низкие энергетические длины волн излучения и малые нулевые энергии возбуждения по сравнению с комплексами иридия. Фактически, хотя комплексы рутения в основном состоянии могут быть сильными восстановителями, комплекс в возбужденном состоянии является гораздо менее сильным восстановителем или окислителем, чем его эквивалентный комплекс иридия. Это делает иридий предпочтительным для развития общих органических превращений, поскольку более сильные окислительно-восстановительные потенциалы возбужденного катализатора позволяют использовать более слабые стехиометрические восстановители и окислители или использовать менее реакционноспособные субстраты. [ 4 ]
Противоионная идентичность
Часто эти фотокатализаторы уравновешиваются противоионом, как в случае с примером комплекса трис-(2,2'-бипиридил)рутения , который сопровождается двумя анионами для уравновешивания общего заряда ионной пары. до нуля. Однако существуют фотоокислительно-восстановительные катализаторы на основе переходных металлов, которые существуют без противоиона, такие как трис(2-фенилпиридин)иридий (часто сокращенно Ir(ppy) 3 ). Значение этих противоионов зависит от ионной ассоциации между фотоокислительно-восстановительным катализатором и его противоионом(ами) и зависит от растворителя , используемого для реакции. Хотя фотофизические свойства, такие как окислительно-восстановительный потенциал, энергия возбуждения и электроотрицательность лиганда, часто считаются ключевыми параметрами использования и реакционной способности этих комплексов, было показано, что идентичность противоиона играет значительную роль в низкополярных растворителях . [ 5 ] [ 6 ] В частности, было показано, что наличие тесно связанного противоиона увеличивает скорость переноса электронов при восстановлении субстрата, но значительно снижает скорость переноса электронов при окислении субстрата. Считается, что это происходит потому, что противоион по существу «блокирует» перенос электронов в фотоокислительно-восстановительный комплекс, экранируя более положительно заряженную область комплекса; тогда как наличие плотной ассоциации противоионов отодвигает электронную плотность дальше от металлического центра фотоокислительно-восстановительного катализатора, облегчая ее перенос из катализатора (конечно, это применимо только к тому случаю, когда фотоокислительно-восстановительный катализатор представляет собой катион , а противоион -ион – анион ). Таким образом, идентичность противоиона является дополнительным параметром, который следует учитывать при разработке новых фотоокислительно-восстановительных реакций.
Приложения
[ редактировать ]Восстановительное дегалогенирование
[ редактировать ]Самое раннее применение фотоокислительно-восстановительного катализа для восстановительного дегалогенирования было ограничено узким диапазоном субстратов или конкурирующим восстановительным сочетанием. [ 7 ]
Неактивированные связи углерод-йод можно восстановить с помощью сильно восстанавливающего фотокатализатора трис-(2,2'- фенилпиридин )иридия (Ir(ppy) 3 ). [ 8 ] Повышенный восстановительный потенциал Ir(ppy) 3 по сравнению с [Ru(bipy) 3 ] 2+ позволяет напрямую восстанавливать связь углерод-йод без взаимодействия со стехиометрическим восстановителем. Таким образом, комплекс иридия передает электрон подложке, вызывая фрагментацию подложки и окисляя катализатор до степени окисления Ir(IV). Окисленный фотокатализатор возвращается в исходное состояние окисления за счет окисления реакционных добавок.

Подобно реакциям радикального дегалогенирования с участием олова, фотокаталитическое восстановительное дегалогенирование может использоваться для инициирования каскадных циклизаций. [ 9 ]

Окислительная генерация ионов иминия
[ редактировать ]иминия Ионы являются мощными электрофилами, полезными для образования связей CC в сложных молекулах. Однако конденсация аминов с карбонильными соединениями с образованием иминий-ионов часто бывает неблагоприятной, иногда требуя жестких условий обезвоживания. Таким образом, альтернативные методы генерации ионов иминия, в частности путем окисления соответствующего амина, являются ценным инструментом синтеза. Ионы иминия можно получить из активированных аминов, используя Ir(dtbbpy)(ppy) 2 PF 6 в качестве фотоокислительно-восстановительного катализатора. [ 10 ] Предполагается, что это превращение происходит путем окисления амина до аминия -радикала катион возбужденным фотокатализатором. После этого происходит перенос атома водорода на сверхстехиметрический окислитель, например трихлорметильный радикал (CCl 3 с образованием иминиевого иона). Затем ион иминия гасится реакцией с нуклеофилом. родственные превращения аминов с широким спектром других нуклеофилов Были исследованы , таких как цианиды ( реакция Стрекера ), силиленольные эфиры ( реакция Манниха ), диалкилфосфаты, аллилсиланы ( реакция аза-Сакураи ), индолы ( реакция Фриделя-Крафтса ), и ацетилиды меди. [ 11 ] [ 12 ] [ 13 ] [ 14 ] [ 15 ]

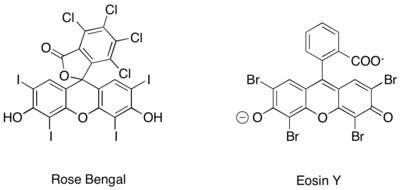
Подобное фотоокислительно-восстановительное генерирование ионов иминия, кроме того, было достигнуто с использованием чисто органических фотоокислительно-восстановительных катализаторов, таких как бенгальский розовый и эозин Y. [ 16 ] [ 17 ] [ 18 ]
В асимметричном варианте этой реакции используются эквиваленты ацил-нуклеофлов, генерируемые катализом N-гетероциклических карбенов . [ 19 ] Этот метод реакции позволяет обойти проблему плохой энантиоиндукции хиральных фотоокислительно-восстановительных катализаторов за счет перемещения источника энантиоселективности в N-гетероциклический карбен.
Окислительная генерация ионов оксокарбения
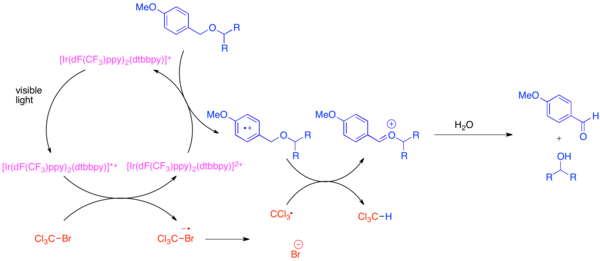
[ редактировать ]Разработка ортогональных защитных групп является проблемой в органическом синтезе, поскольку эти защитные группы позволяют каждый экземпляр общей функциональной группы, такой как гидроксильная различить группа, во время синтеза сложной молекулы. Очень распространенной защитной группой для гидроксильной функциональной группы является пара -метоксибензиловый (ПМБ) эфир. Эта защитная группа химически подобна менее богатому электронами бензиловому эфиру. Обычно для селективного расщепления эфира ПМБ в присутствии бензилового эфира используются сильные стехиометрические окислители, такие как 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ) или нитрат церия-аммония (CAN). Эфиры ПМБ гораздо более восприимчивы к окислению, чем бензиловые эфиры, поскольку они более богаты электронами. Селективное снятие защиты с эфиров ПМБ можно достичь за счет использования гексафторфосфата бис-(2-(2',4'-дифторфенил)-5-трифторметилпиридин)-(4,4'-дитретбутилбипиридин)иридия(III) (Ir[dF (CF 3 )ppy] 2 (dtbbpy)PF 6 ) и мягкий стехиометрический окислитель, такой как бромтрихлорметан, BrCCl 3 . [ 20 ] Фотовозбужденный иридиевый катализатор восстанавливается настолько, что фрагментирует бромтрихлорметан с образованием трихлорметильного радикала, бромид-аниона и комплекса Ir(IV). Бедные электронами фторированные лиганды делают иридиевый комплекс достаточно окисляющим, чтобы принять электрон от богатого электронами арена, такого как эфир ПМБ. После окисления арена он легко участвует в переносе атома водорода с трихлорметильным радикалом с образованием хлороформа и иона оксокарбения , который легко гидролизуется с образованием свободного гидроксида. Было показано, что эта реакция ортогональна многим обычным защитным группам, когда для нейтрализации образующегося HBr добавлялось основание.
Циклодополнения
[ редактировать ]Циклоприсоединения и другие перициклические реакции являются мощными преобразованиями в органическом синтезе из-за их способности быстро создавать сложную молекулярную архитектуру и, в частности, из-за их способности множество соседних стереоцентров жестко контролировать . Однако в термических условиях разрешены только определенные циклоприсоединения в соответствии с правилами орбитальной симметрии Вудворда-Хоффмана или другими эквивалентными моделями, такими как теория пограничных молекулярных орбиталей (FMO) или модель Дьюара-Циммермана. Циклоприсоединения, которые не разрешены термически, такие как [2+2] циклоприсоединение, могут быть активированы фотохимической активацией реакции. В некаталитических условиях эта активация требует использования ультрафиолетового света высокой энергии , способного изменять орбитальные популяции реакционноспособных соединений. Альтернативно, сообщалось, что металлические катализаторы, такие как кобальт и медь, катализируют термически запрещенное [2+2] циклоприсоединение посредством переноса одного электрона.

Требуемое изменение орбитальной населенности может быть достигнуто путем переноса электронов с помощью фотокатализатора, чувствительного к видимому свету более низкой энергии. [ 21 ] [ 22 ] [ 23 ] [ 24 ] [ 25 ] Юн продемонстрировал эффективное внутри- и межмолекулярное [2+2] циклоприсоединение активированных олефинов , особенно енонов и стиролов. Было обнаружено, что еноны, или бедные электронами олефины, реагируют по радикально-анионному пути, используя диизопропилэтиламин в качестве временного источника электронов. Для этого переноса электрона [Ru(bipy) 3 ] 2+ было обнаружено, что он является эффективным фотокатализатором. Анионная природа циклизации оказалась решающей: проведение реакции в кислоте, а не с противоионом лития, способствовало пути нециклоприсоединения. [ 26 ] Чжао и др. доступен еще один путь циклизации аналогичным образом обнаружил, что халконам с противоионом самария . [ 27 ] И наоборот, было обнаружено, что богатые электронами стиролы реагируют по радикально-катионному механизму, используя метилвиологен или молекулярный кислород в качестве временного поглотителя электронов. Пока [Ru(bipy) 3 ] 2+ оказался компетентным катализатором внутримолекулярной циклизации с использованием метилвиологена , его нельзя было использовать с молекулярным кислородом в качестве стока электронов или для межмолекулярной циклизации. Что касается межмолекулярной циклизации, Yoon et al. обнаружили, что более сильно окисляющий фотокатализатор [Ru(bpm) 3 ] 2+ и молекулярный кислород обеспечил каталитическую систему, лучше подходящую для доступа к катион-радикалу, необходимому для возникновения циклоприсоединения. [Ру(бпз) 3 ] 2+ , еще более сильно окисляющий фотокатализатор, оказался проблематичным, поскольку, хотя он мог катализировать желаемое [2+2] циклоприсоединение, он также был достаточно сильным, чтобы окислить циклоаддукт и катализировать ретро-[2+2] реакцию. Это сравнение фотокатализаторов подчеркивает важность настройки окислительно-восстановительных свойств фотокатализатора в соответствии с реакционной системой, а также демонстрирует ценность полипиридильных соединений в качестве лигандов из-за легкости, с которой их можно модифицировать для регулировки окислительно-восстановительных свойств их комплексов.

Фоторедокс-катализируемое [2+2]-циклоприсоединение также можно осуществлять с помощью трифенилпирилиевого органического фотоокислительно-восстановительного катализатора. [ 28 ]

Помимо термически запрещенного [2+2] циклоприсоединения, фотоокислительно-восстановительный катализ может быть применен к циклизации [4+2] ( реакция Дильса-Альдера ). Бис-еноны, подобные субстратам, используемым для фоторедокс-[2+2]-циклизации, но с более длинным линкером, соединяющим две еноновые функциональные группы, вступают в внутримолекулярные радикально-анионные гетеро-реакции Дильса-Альдера быстрее, чем [2+2] циклоприсоединение. [ 29 ]

Аналогично, богатые электронами стиролы участвуют во внутри- или межмолекулярных циклизациях Дильса-Альдера по катион-радикальному механизму. [ 30 ] [ 31 ] [Ru(bipy) 3 ] 2+ был компетентным катализатором межмолекулярной, но не внутримолекулярной циклизации Дильса-Альдера. Эта фоторедокс-катализируемая реакция Дильса-Альдера допускает циклоприсоединение между двумя электронно несогласованными субстратами. Обычный электронный запрос для реакции Дильса-Альдера требует, чтобы богатый электронами диен реагировал с бедным электронами олефином (или «диенофилом»), в то время как реакция Дильса-Альдера с обратным электронным запросом происходит между противоположным случаем бедный электронами диен и очень богатый электронами диенофил. Фоторедокс-случай, поскольку он происходит по иному механизму, чем термическая реакция Дильса-Альдера, допускает циклоприсоединение между богатым электронами диеном и богатым электронами диенофилом, обеспечивая доступ к новым классам аддуктов Дильса-Альдера.

Синтетическая ценность фоторедокс-катализируемой Юном стирольной реакции Дильса-Альдера была продемонстрирована посредством полного синтеза природного продукта гейциамида А. [ 30 ] Этот синтез демонстрирует, что термическая реакция Дильса-Альдера благоприятствует нежелательному региоизомеру, но фоторедокс-катализируемая реакция дает желаемый региоизомер с повышенным выходом.

Фотоокислительно-восстановительный органокатализ
[ редактировать ]Органокатализ - это подобласть катализа, которая исследует потенциал органических малых молекул в качестве катализаторов, особенно для энантиоселективного создания хиральных молекул. Одной из стратегий в этой области является использование хиральных вторичных аминов для активации карбонильных соединений. В этом случае конденсация амина с карбонильным соединением приводит к образованию нуклеофильного енамина . Хиральный амин устроен так, что одна сторона енамина стерически экранирована и только неэкранированная сторона может реагировать. Несмотря на способность этого подхода катализировать энантиоселективную функционализацию карбонильных соединений, некоторые ценные превращения, такие как каталитическое энантиоселективное α-алкилирование альдегидов , оставались неуловимыми. Сочетание органокатализа и фотоокислительно-восстановительных методов обеспечивает каталитическое решение этой проблемы. [ 32 ] В этом подходе для α-алкилирования альдегидов [Ru(bipy) 3 ] 2+ восстановительно фрагментирует активированный алкилгалогенид, такой как бромомалонат или фенацилбромид , который затем может быть добавлен к каталитически полученному енамину энантиоселективным образом. Окисленный фотокатализатор затем окислительно тушит образовавшийся α-аминорадикал с образованием иона иминия, который гидролизуется с образованием функционализированного карбонильного соединения. Было показано, что это фотоокислительно-восстановительное превращение механически отличается от другого органокаталитического радикального процесса, называемого катализом на однозанятой молекулярной орбитали (SOMO). В катализе SOMO используется сверхстехиометрический нитрат церия-аммония (CAN) для окисления каталитически полученного енамина до соответствующего катион-радикала, который затем может быть добавлен к подходящему партнеру сочетания, такому как аллилсилан. Этот тип механизма исключен для реакции фотокаталитического алкилирования, поскольку в то время как катион-радикал енамина наблюдался циклизацией на подвесных олефинах и открытых циклопропановых радикальных часах в катализе SOMO, эти структуры были нереакционноспособны в фотоокислительно-восстановительной реакции.
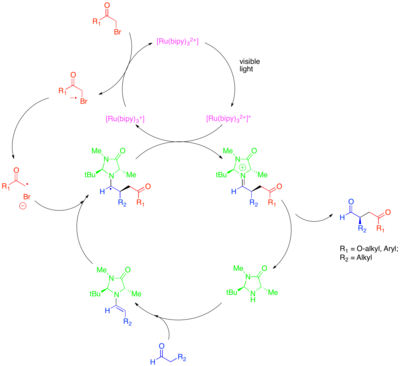
Это преобразование включает алкилирование другими классами активированных алкилгалогенидов, представляющих синтетический интерес. В частности, использование фотокатализатора Ir(dtbbpy)(ppy) 2 + позволяет энантиоселективное α-трифторметилирование альдегидов, тогда как использование Ir(ppy) 3 позволяет энантиоселективное сочетание альдегидов с бедными электронами бензилбромидами. [ 33 ] [ 34 ] Цейтлер и др. также исследовал продуктивное слияние фотоокислительно-восстановительных и органокаталитических методов для достижения энантиоселективного алкилирования альдегидов. [ 35 ] Тот же хиральный имидазолидиноновый органокатализатор использовали для образования енамина и введения хиральности. Однако вместо комплекса рутения или иридия использовался органический фотоокислительно-восстановительный катализатор эозин Y.
Прямое β-арилирование насыщенных альдегидов и кетонов можно осуществлять сочетанием фотоокислительно-восстановительных и органокаталитических методов. [ 36 ] Предыдущий метод осуществления прямой β-функционализации насыщенного карбонила состоит из одного процесса, состоящего из двух стадий, оба из которых катализируются вторичным аминным органокатализатором: стехиометрическое восстановление альдегида с помощью IBX с последующим добавлением активированного алкилнуклеофила. в бета-положение полученного энала . [ 37 ] Это превращение, протекающее, как и другие фотоокислительно-восстановительные процессы, по радикальному механизму, ограничивается присоединением высокоэлектрофильных аренов в бета-положение. Серьезные ограничения на количество ареновых компонентов в этой реакции обусловлены, прежде всего, необходимостью в анион-радикале арен, который достаточно стабилен, чтобы не реагировать напрямую с енамином или катион-радикалом енамина. В предлагаемом механизме активированный фотоокислительно-восстановительный катализатор гасится окислительно электронодефицитным ареном, таким как 1,4-дицианобензол . Затем фотокатализатор окисляет разновидность енамина, временно образующуюся в результате конденсации альдегида с сокатализатором вторичного амина, таким как оптимальный изопропилбензиламин. Образующийся катион-радикал енамина обычно реагирует как 3 π-электронная система, но из-за стабильности партнеров радикального взаимодействия депротонирование β-метиленовой позиции приводит к образованию 5 π-электронной системы с сильным радикальным характером во вновь доступном месте. β-углерод. Хотя эта реакция основана на использовании вторичного аминного органокатализатора для образования енаминов, которые окисляются по предлагаемому механизму, энантиоселективного варианта этой реакции не существует.

Развитие этого прямого β-арилирования альдегидов привело к родственным реакциям β-функционализации циклических кетонов. В частности, β-арилирование циклических кетонов было достигнуто в аналогичных условиях реакции, но с использованием азепана в качестве сокатализатора вторичного амина. Фотокаталитическая «гомо-альдольная» реакция работает с циклическими кетонами, позволяя соединить бета-положение кетона с ипсо-углеродом арилкетонов, таких как бензофенон и ацетофенон . [ 38 ] Помимо азепанового сокатализатора, эта реакция требует использования более сильно восстанавливающего фотоокислительно-восстановительного катализатора Ir(ppy) 3 и добавления гексафторарсенида лития (LiAsF 6 ) для содействия одноэлектронному восстановлению арилкетона.
Добавки к олефинам
[ редактировать ]Использование фотоокислительно-восстановительного катализа для генерации реакционноспособных гетероатом-центрированных радикалов было впервые исследовано в 1990-х годах. [ 39 ] [Ru(bipy) 3 ] 2+ Было обнаружено, что он катализирует фрагментацию тозилфенилселенида на фенилселенат-анион и тозильный радикал, и что механизм развития радикальной цепи позволяет присоединять тозильный радикал и фенилселенорадикал через двойную связь богатых электронами алкилвиниловых эфиров. Поскольку фенилселенят-анион легко окисляется до дифенилдиселенида, наблюдаемые низкие количества дифенилдиселенида были восприняты как указание на то, что фоторедокс-катализируемая фрагментация тозилфенилселенида была важна только как стадия инициирования и что большая часть реакционной способности была обусловлена радикально-цепным процессом.
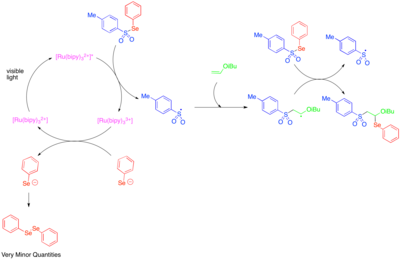
Гетероароматические присоединения к олефинам включают реакции многокомпонентного окси- и аминотрифторметилирования. [ 40 ] [ 41 ] В этих реакциях используется реагент Умемото, сульфониевая соль, которая служит электрофильным источником трифторметильной группы и, как известно, реагирует по пути одноэлектронного переноса. Таким образом, одноэлектронное восстановление реагента Умемото высвобождает трифторметильный радикал, который присоединяется к реакционноспособному олефину. Впоследствии одноэлектронное окисление алкильного радикала, образующегося в результате этого присоединения, приводит к образованию катиона, который может быть захвачен водой, спиртом или нитрилом. Чтобы достичь высокого уровня региоселективности, эта реакционная способность исследовалась в основном для стиролов, которые склонны к образованию промежуточного бензильного радикала.

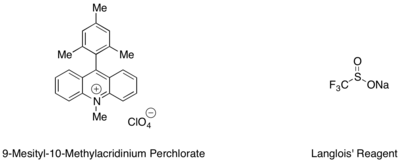
Гидротрифторметилирование стиролов и алифатических алкенов можно осуществлять с использованием мезитилакридиниевого органического фотоокислительно-восстановительного катализатора и реактива Ланглуа в качестве источника радикала CF 3 . [ 42 ] В этой реакции было обнаружено, что трифторэтанол и субстехиометрические количества ароматического тиола, такого как метилтиосалицилат, используемые совместно, служат лучшим источником водородного радикала для завершения каталитического цикла.
Внутримолекулярные гидроэтерификации и гидроаминирования протекают с антимарковниковской селективностью. [ 43 ] [ 44 ] Один механизм включает одноэлектронное окисление олефина, захватывание катион-радикала боковой гидроксильной или аминной функциональной группой и гашение образующегося алкильного радикала путем переноса атома водорода от высоколабильных донорных частиц. Распространение этой реакционной способности на межмолекулярные системы привело к i) новому пути синтеза сложных тетрагидрофуранов путем «циклоприсоединения с перекрестным полярным радикалом» (реакция PRCC) аллильного спирта с олефином и ii) антимарковниковского добавления карбоновые кислоты в олефины. [ 45 ] [ 46 ]
Сульфоксимидирование
[ редактировать ]Недавно сообщалось о поздней стадии сульфоксимидирования богатых электронами аренов посредством фотоокислительно-восстановительного катализа и обсуждалось несколько примеров. [ 47 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Ромеро, Натан А.; Ницевич, Дэвид А. (10 июня 2016 г.). «Органический фотоокислительно-восстановительный катализ». Химические обзоры . 2016 (116): 10075–10166. doi : 10.1021/acs.chemrev.6b00057 . ПМИД 27285582 .
- ^ Джонс, Уэйн Э.; Фокс, Мэри Энн (май 1994 г.). «Определение окислительно-восстановительных потенциалов возбужденного состояния методом фазово-модулированной вольтамперометрии». Журнал физической химии . 98 (19): 5095–5099. дои : 10.1021/j100070a025 .
- ^ «Электрохимический ряд фотокатализаторов и обычных органических соединений» (PDF) . Мерк . Проверено 15 апреля 2019 г.
- ^ Jump up to: а б Такер, Джозеф В.; Стивенсон, Кори Р.Дж. (2012). «Проливающий свет на фотоокислительно-восстановительный катализ: теория и синтетические приложения». Журнал органической химии . 77 (4): 1617–1622. дои : 10.1021/jo202538x . ПМИД 22283525 .
- ^ Эрли, доктор медицинских наук; Зеленевская, А.; Рипбергер, Х.Х.; Шин, Нью-Йорк; Лазорский, М.С.; Маст, З.Дж.; Сэйр, HJ; Маккаскер, Дж. К.; Скоулз, Джорджия; Ноулз, Р.Р.; Рид, Огайо (14 апреля 2022 г.). «Реорганизация ионных пар регулирует реакционную способность фотоокислительно-восстановительных катализаторов» . Природная химия . 14 (7): 746–753. Бибкод : 2022NatCh..14..746E . дои : 10.1038/s41557-022-00911-6 . ISSN 1755-4349 . ПМИД 35422457 . S2CID 248152234 .
- ^ Фарни, Эллиот П.; Чепмен, Стивен Дж.; Сордс, Уэсли Б.; Торелли, Марко Д.; Хамерс, Роберт Дж.; Юн, Техшик П. (17 апреля 2019 г.). «Открытие и выяснение контранионной зависимости в фотоокислительно-восстановительном катализе» . Журнал Американского химического общества . 141 (15): 6385–6391. дои : 10.1021/jacs.9b01885 . ISSN 0002-7863 . ПМК 6519111 . ПМИД 30897327 .
- ^ Нараянам, Джаган М.Р.; Джозеф В. Такер; Кори Р. Дж. Стивенсон (5 июня 2009 г.). «Фотоокислительно-восстановительный катализ с переносом электрона: разработка процедуры восстановительного дегалогенирования без олова». Журнал Американского химического общества . 131 (25): 8756–8757. дои : 10.1021/ja9033582 . ПМИД 19552447 .
- ^ Нгуен, Джон Д.; Д'Амато, Эрика М.; Нараянам, Джаган М.Р.; Стивенсон, Кори Р.Дж. (2012). «Вовлечение неактивированных алкилов, алкенилов и арилйодидов в свободнорадикальные реакции, опосредованные видимым светом». Природная химия . 4 (10): 854–859. Бибкод : 2012НатЧ...4..854Н . дои : 10.1038/nchem.1452 . ПМИД 23001000 . S2CID 29747574 .
- ^ Ферст, Лаура; Нараянам, Джаган М.Р.; Стивенсон, Кори Р.Дж. (4 октября 2011 г.). «Полный синтез (+)-глиокладина C с помощью фотоокислительно-восстановительного катализа в видимом свете» . Angewandte Chemie, международное издание . 50 (41): 9655–9659. дои : 10.1002/anie.201103145 . ПМК 3496252 . ПМИД 21751318 .
- ^ Конди, Эллисон Г.; Гонсалес-Гомес, Хосе К.; Стивенсон, Кори Р.Дж. (10 февраля 2010 г.). «Фотоокислительно-восстановительный катализ в видимом свете: реакции аза-Генри посредством функционализации CH». Журнал Американского химического общества . 132 (5): 1464–1465. дои : 10.1021/ja909145y . ПМИД 20070079 .
- ^ Рюпинг, Магнус; Чжу, Шаоцюнь; Кенигс, Рене М. (2011). «Фотоокислительно-восстановительная реакция, катализируемая видимым светом, окислительная реакция Стрекера». Химические коммуникации . 47 (47): 12709–11. дои : 10.1039/C1CC15643H . ПМИД 22041859 .
- ^ Чжао, Гуолей; Ян, Чао; Го, Линь; Сунь, Хуннань; Чен, Чао; Ся, Уцзюн (2012). «Реакция окислительного сочетания, индуцированная видимым светом: легкий доступ к продуктам типа Манниха». Химические коммуникации . 48 (17): 2337–9. дои : 10.1039/C2CC17130A . ПМИД 22252544 .
- ^ Рюпинг, Магнус; Чжу, Шаоцюнь; Кенигс, Рене М. (2011). «Фоторедокс-катализируемые реакции образования связи C–P - окислительное фосфонилирование аминов, опосредованное видимым светом». Химические коммуникации . 47 (30): 8679–81. дои : 10.1039/C1CC12907D . ПМИД 21720622 .
- ^ Фриман, Дэвид Б.; Ферст, Лаура; Конди, Эллисон Г.; Стивенсон, Кори Р.Дж. (6 января 2012 г.). «Функционально разнообразный нуклеофильный захват промежуточных соединений иминия, полученный с использованием видимого света» . Органические письма . 14 (1): 94–97. дои : 10.1021/ol202883v . ПМК 3253246 . ПМИД 22148974 .
- ^ Рюпинг, Магнус; Кенигс, Рене М.; Пошарный Константин; Фабри, Дэвид С.; Леонори, Даниэле; Вила, Карлос (23 апреля 2012 г.). «Двойной катализ: сочетание фотокаталитического аэробного окисления и катализируемых металлами реакций алкинилирования - образование связи C≡C с использованием видимого света». Химия: Европейский журнал . 18 (17): 5170–5174. дои : 10.1002/chem.201200050 . ПМИД 22431393 .
- ^ Пан, Юаньхан; Ван, Шуай; Ки, Чун Ви; Дюбюиссон, Эмили; Ян, Юаньён; Ло, Киан Пинг; Тан, Чун-Хонг (2011). «Оксид графена и бенгальская роза: окислительная C–H-функционализация третичных аминов с использованием видимого света». Зеленая химия . 13 (12): 3341. doi : 10.1039/C1GC15865A .
- ^ Фу, Вэйцзюнь; Го, Вэньбо; Цзоу, Гуанлун; Сюй, Чен (август 2012 г.). «Селективное трифторметилирование и алкинилирование тетрагидроизохинолинов с использованием облучения видимым светом бенгальской розы». Журнал химии фтора . 140 : 88–94. дои : 10.1016/j.jfluchem.2012.05.009 .
- ^ Хари, Дурга Прасад; Кениг, Буркхард (5 августа 2011 г.). «Образование связей C–C и C–P, катализируемое видимым светом эозином Y». Органические письма . 13 (15): 3852–3855. дои : 10.1021/ol201376v . ПМИД 21744842 .
- ^ ДиРокко, Дэниел А.; Ровис, Томислав (16 мая 2012 г.). «Каталитическое асимметричное α-ацилирование третичных аминов, опосредованное двойным режимом катализа: N-гетероциклическим карбеном и фотоокислительно-восстановительным катализом» . Журнал Американского химического общества . 134 (19): 8094–8097. дои : 10.1021/ja3030164 . ПМК 3354013 . ПМИД 22548244 .
- ^ Такер, Джозеф В.; Нараянам, Джаган М.Р.; Шах, Пинки С.; Стивенсон, Кори Р.Дж. (2011). «Окислительный фотоокислительно-восстановительный катализ: мягкое и селективное снятие защиты с эфиров ПМБ, опосредованное видимым светом». Химические коммуникации . 47 (17): 5040–5042. дои : 10.1039/c1cc10827a . ПМИД 21431223 .
- ^ Ишай, Майкл А.; Анзовино, Мэри Э.; Ду, Хуана; Юн, Техшик П. (октябрь 2008 г.). «Эффективный фотокатализ [2+2] еноновых циклоприсоединений в видимом свете». Журнал Американского химического общества . 130 (39): 12886–12887. дои : 10.1021/ja805387f . ПМИД 18767798 .
- ^ Ду, Хуана; Юн, Техшик П. (21 октября 2009 г.). «Скрещенные межмолекулярные [2+2] циклоприсоединения ациклических енонов посредством фотокатализа в видимом свете» . Журнал Американского химического общества . 131 (41): 14604–14605. дои : 10.1021/ja903732v . ПМК 2761970 . ПМИД 19473018 .
- ^ Ишай, Майкл А.; Лу, Жан; Юн, Техшик П. (30 июня 2010 г.). «[2+2] Циклоприсоединения посредством окислительного фотокатализа в видимом свете» . Журнал Американского химического общества . 132 (25): 8572–8574. дои : 10.1021/ja103934y . ПМЦ 2892825 . ПМИД 20527886 .
- ^ Тайсон, Элизабет Л.; Фарни, Эллиот П.; Юн, Техшик П. (17 февраля 2012 г.). «Фотокаталитическое [2 + 2] циклоприсоединение енонов с расщепляемыми окислительно-восстановительными вспомогательными веществами» . Органические письма . 14 (4): 1110–1113. дои : 10.1021/ol3000298 . ПМЦ 3288794 . ПМИД 22320352 .
- ^ Ишай, Майкл А.; Амент, Майкл С.; Юн, Техшик П. (2012). «Скрещенное межмолекулярное [2 + 2] циклоприсоединение стирола методом фотокатализа в видимом свете» . Химическая наука . 3 (9): 2807–2811. дои : 10.1039/c2sc20658g . ПМЦ 3439822 . ПМИД 22984640 .
- ^ Ду, Хуана; Эспельт, Лаура Руис; Гузей Илья А.; Юн, Техшик П. (2011). «Фотокаталитическая восстановительная циклизация енонов: разная реакционная способность фотогенерированных радикалов и промежуточных анионных радикалов» . Химическая наука . 2 (11): 2115–2119. дои : 10.1039/c1sc00357g . ПМК 3222952 . ПМИД 22121471 .
- ^ Чжао, Гуолей; Ян, Чао; Го, Линь; Сунь, Хуннань; Лин, Беги; Ся, Уцзюн (20 июля 2012 г.). «Понимание реакционной способности восстановительного взаимодействия и альдольной циклизации халконов с помощью фотокатализа в видимом свете». Журнал органической химии . 77 (14): 6302–6306. дои : 10.1021/jo300796j . ПМИД 22731518 .
- ^ Ринер, Мишель; Ницевич, Дэвид А. (2013). «Синтез циклобутан-лигнанов с помощью органической одноэлектронной системы окислитель – электронная релейная система» . Химическая наука . 4 (6): 2625. дои : 10.1039/c3sc50643f . ПМЦ 3862357 . ПМИД 24349680 .
- ^ Хертли, Анна Э.; Чисмезия, Меган А.; Ишай, Майкл А.; Юн, Техшик П. (июнь 2011 г.). «Видимый световой фотокатализ анион-радикальных гетеро-циклоприсоединения Дильса – Альдера» . Тетраэдр . 67 (24): 4442–4448. дои : 10.1016/j.tet.2011.02.066 . ПМК 3110713 . ПМИД 21666769 .
- ^ Jump up to: а б Лин, Шиши; Ишай, Майкл А.; Фрай, Чарльз Г.; Юн, Техшик П. (7 декабря 2011 г.). «Катион-радикалы Дильса – Циклоприсоединения ольхи с помощью фотокатализа в видимом свете» . Журнал Американского химического общества . 133 (48): 19350–19353. дои : 10.1021/ja2093579 . ПМЦ 3227774 . ПМИД 22032252 .
- ^ Лин, Шиши; Падилья, Кристиан Э.; Ишай, Майкл А.; Юн, Техшик П. (июнь 2012 г.). «Видимый световой фотокатализ внутримолекулярных катион-радикалов циклоприсоединения Дильса – Альдера» . Буквы тетраэдра . 53 (24): 3073–3076. дои : 10.1016/j.tetlet.2012.04.021 . ПМЦ 3375996 . ПМИД 22711942 .
- ^ Ницевич, Д.А.; Макмиллан, DWC (3 октября 2008 г.). «Объединение фотоокислительно-восстановительного катализа с органокализом: прямое асимметричное алкилирование альдегидов» . Наука . 322 (5898): 77–80. Бибкод : 2008Sci...322...77N . дои : 10.1126/science.1161976 . ПМЦ 2723798 . ПМИД 18772399 .
- ^ Нагиб, Дэвид А.; Скотт, Марк Э.; Макмиллан, Дэвид У.К. (12 августа 2009 г.). «Энантиоселективное α-трифторметилирование альдегидов посредством фотоокислительно-восстановительного органокатализа» . Журнал Американского химического общества . 131 (31): 10875–10877. дои : 10.1021/ja9053338 . ПМК 3310169 . ПМИД 19722670 .
- ^ Ши, Хуэй-Вэнь; Вандер Уол, Марк Н.; Грейндж, Ребекка Л.; Макмиллан, Дэвид У.К. (6 октября 2010 г.). «Энантиоселективное α-бензилирование альдегидов посредством фотоокислительно-восстановительного органокатализа» . Журнал Американского химического общества . 132 (39): 13600–13603. дои : 10.1021/ja106593m . ПМК 3056320 . ПМИД 20831195 .
- ^ Нойманн, Матиас; Фюльднер, Стефан; Кениг, Буркхард; Цейтлер, Кирстен (24 января 2011 г.). «Безметалловый кооперативный асимметричный органофотоокислительно-восстановительный катализ в видимом свете». Международное издание «Прикладная химия» . 50 (4): 951–954. дои : 10.1002/anie.201002992 . ПМИД 20878819 .
- ^ Пирнот, Монтана; Ранник, Д.А.; Мартин, DBC; Макмиллан, DWC (28 марта 2013 г.). «Фоторедокс-активация для прямого арилирования кетонов и альдегидов» . Наука . 339 (6127): 1593–1596. дои : 10.1126/science.1232993 . ПМЦ 3723331 . ПМИД 23539600 .
- ^ Чжан, Ши-Лэй; Се, Хэ-Синь; Чжу, Цзинь; Ли, Хао; Чжан, Синь-Шуай; Ли, Цзянь; Ван, Вэй (1 марта 2011 г.). «Органокаталитическая энантиоселективная β-функционализация альдегидов путем окисления енаминов и их применение в каскадных реакциях» . Природные коммуникации . 2 : 211. Бибкод : 2011NatCo...2..211Z . дои : 10.1038/ncomms1214 . ПМИД 21364550 .
- ^ Петрониевич, Филип Р.; Наппи, Мануэль; Макмиллан, Дэвид У.К. (22 ноября 2013 г.). «Прямая β-функционализация циклических кетонов с помощью арилкетонов посредством слияния фотоокислительно-восстановительного процесса и органокатализа» . Журнал Американского химического общества . 135 (49): 18323–18326. дои : 10.1021/ja410478a . ПМЦ 3934322 . ПМИД 24237366 .
- ^ Бартон, Дерек HR; Чиба, Мэри А.; Ясбереньи, Джозеф Ч. (май 1994 г.). "Ру(bpy ) 2+ -опосредованное присоединение Se-фенил-п-толуолселеносульфоната к олефинам, богатым электронами». Tetrahedron Letters . 35 (18): 2869–2872. doi : 10.1016/S0040-4039(00)76646-9 .
- ^ Ясу, Юсуке; Койке, Такаши; Акита, Мунетака (17 сентября 2012 г.). «Трехкомпонентное окситрифторметилирование алкенов: высокоэффективная и региоселективная дифункционализация связей C=C, опосредованная фотоокислительно-восстановительными катализаторами». Angewandte Chemie, международное издание . 51 (38): 9567–9571. дои : 10.1002/anie.201205071 . ПМИД 22936394 .
- ^ Ясу, Юсуке; Койке, Такаши; Акита, Мунетака (3 мая 2013 г.). «Межмолекулярное аминотрифторметилирование алкенов посредством фотоокислительно-восстановительного катализа, управляемого видимым светом». Органические письма . 15 (9): 2136–2139. дои : 10.1021/ol4006272 . ПМИД 23600821 .
- ^ Уилгер, Дейл Дж.; Джесмундо, Натан Дж.; Ницевич, Дэвид А. (2013). «Каталитическое гидротрифторметилирование стирола и неактивированных алифатических алкенов с помощью органической фотоокислительно-восстановительной системы». Химическая наука . 4 (8): 3160. дои : 10.1039/c3sc51209f .
- ^ Гамильтон, Дэвид С.; Ницевич, Дэвид А. (14 ноября 2012 г.). «Прямая каталитическая антимарковниковская гидроэтерификация алкенолов» . Журнал Американского химического общества . 134 (45): 18577–18580. дои : 10.1021/ja309635w . ПМЦ 3513336 . ПМИД 23113557 .
- ^ Нгуен, Тьен М.; Ницевич, Дэвид А. (3 июля 2013 г.). «Антимарковниковское гидроаминирование алкенов, катализируемое органической фотоокислительно-восстановительной системой» . Журнал Американского химического общества . 135 (26): 9588–9591. дои : 10.1021/ja4031616 . ПМЦ 3754854 . ПМИД 23768239 .
- ^ Гранжан, Жан-Марк М.; Ницевич, Дэвид А. (2 апреля 2013 г.). «Синтез высокозамещенных тетрагидрофуранов каталитическим полярно-радикальным кроссоверным циклоприсоединением алкенов и алкенолов» . Angewandte Chemie, международное издание . 52 (14): 3967–3971. дои : 10.1002/anie.201210111 . ПМИД 23440762 .
- ^ Перковски, Эндрю Дж.; Ницевич, Дэвид А. (17 июля 2013 г.). «Прямое каталитическое антимарковниковское присоединение карбоновых кислот к алкенам» . Журнал Американского химического общества . 135 (28): 10334–10337. дои : 10.1021/ja4057294 . ПМЦ 3757928 . ПМИД 23808532 .
- ^ Мейер, Робин; Хог, Дэниел; Ламмерманн, Генриетта; Судау, Александр; Ракл, Дэниел; Вайнманн, Хилмар; Коллинз, Карл; Вортманн, Ларс; Кэндиш, Лиза (2018). «Поздняя стадия сульфоксимидирования богатых электронами аренов с помощью фотоокислительно-восстановительного катализа» . Синлетт . 29 (20): 2679–2684. дои : 10.1055/s-0037-1609656 . S2CID 105213357 .