Хардеропорфирия
| Хардеропорфирия | |
|---|---|
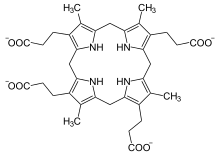 | |
| Копропорфириноген III |
Хардеропорфирия — редкое нарушение биосинтеза гема , наследуемое по аутосомно-рецессивному типу и обусловленное специфическими мутациями гена CPOX . Мутации в CPOX обычно вызывают наследственную копропорфирию , острую печеночную порфирию , однако мутация K404E в гомозиготном или компаунд-гетерозиготном состоянии с нулевым аллелем вызывает более тяжелую хардопорфирию. [ 1 ] Хардеропорфирия — первое известное нарушение обмена веществ, фенотип которого зависит от типа и локализации мутаций в гене, связанном с множественными нарушениями. [ 2 ]
В отличие от других порфирий, которые обычно проявляются либо кожными поражениями после воздействия солнечного света, либо острым нейровисцеральным приступом в любом возрасте (чаще всего в зрелом возрасте), хардопорфирия характеризуется желтухой , анемией, увеличением печени и селезенки , часто проявляющейся в неонатальном периоде. В более позднем возрасте у этих людей может проявляться светочувствительность, аналогичная той, которая наблюдается при кожных порфириях . [ 2 ]
Биохимически хардопорфирия проявляется отчетливой картиной повышенного содержания хардопорфирина (порфирина 2-винил-4,6,7-трипропионовой кислоты). [ 3 ] в моче и особенно в кале — метаболит, который не обнаруживается в значительных количествах при любой другой порфирии. [ 2 ] Ферментные тесты показывают заметно сниженную активность копропорфириногеноксидазы по сравнению как с незараженными людьми, так и с лицами, страдающими наследственной копропорфирией, что соответствует рецессивному наследованию. [ 2 ]
Хардеропорфирия — редкое заболевание: во всем мире зарегистрировано менее 10 случаев. Его может не диагностировать, поскольку он не имеет типичного проявления, связанного с порфирией. [ 2 ] Он был идентифицирован как вариантный тип копропорфирии в 1983 году в семье с тремя детьми, у которых при рождении была выявлена желтуха и гемолитическая анемия. [ 4 ] Стандартного лечения хардопорфирии не существует; помощь в основном сосредоточена на лечении симптомов. [ 4 ] [ 5 ]
Ссылки
[ редактировать ]- ^ Шмитт, К.; Гуя, Л.; Малонова Е.; Ламорил, Дж.; Камадро, Дж. М.; Фламме, М.; Роуз, К.; Люми, С.; Да Силва, В.; Буало, К.; Граншан, Б.; Бомонт, К.; Дейбах, Дж. К.; Пюй, Х. (2005). «Мутации в гене CPO человека предсказывают клиническое проявление либо печеночной наследственной копропорфирии, либо эритропоэтической хардопорфирии». Молекулярная генетика человека . 14 (20): 3089–3098. дои : 10.1093/hmg/ddi342 . ПМИД 16159891 .
- ^ Jump up to: а б с д и «#121300 КОПРОПОРФИРИЯ НАСЛЕДСТВЕННАЯ; ГКП» . Университет Джонса Хопкинса . Проверено 27 мая 2012 г.
- ^ Горчейн, А.; Дантон, М.; Лим, СК (октябрь 2005 г.). «Хардеропорфирин: неправильное название». Биомедицинская хроматография . 19 (8): 565–569. дои : 10.1002/bmc.480 . ПМИД 15678522 .
- ^ Jump up to: а б Нордманн, Ю.; Граншан, Б.; Де Верней, Х.; Фунг, Л.; Картиньи, Б.; Фонтейн, Г. (1983). «Хардеропорфирия: вариант наследственной копропорфирии» . Журнал клинических исследований . 72 (3): 1139–1149. дои : 10.1172/JCI111039 . ПМЦ 1129282 . ПМИД 6886003 .
- ^ Ламорил, Дж.; Пюи, Х.; Гуя, Л.; Росипал, Р.; Да Силва, В.; Граншан, Б.; Фойнт, Т.; Бадер-Мёнье, Б.; Доммерг, JP; Дейбах, Джей-Джей; Нордманн, Ю. (1998). «Неонатальная гемолитическая анемия вследствие наследственной хардопорфирии: клинические характеристики и молекулярная основа». Кровь . 91 (4): 1453–1457. ПМИД 9454777 .