Маннозидоз
| Маннозидоз | |
|---|---|
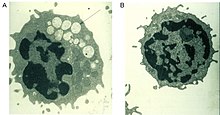 | |
| Электронная микрофотография вакуолизированного лимфоцита пациента с маннозидозом (А) по сравнению с лимфоцитом нормального контроля (Б). | |
| Специальность | Педиатрия , эндокринология |
Маннозидоз это дефицит маннозидазы фермента . – [ 1 ] Существует два типа: альфа-маннозидоз и бета-маннозидоз . Оба расстройства связаны с лизосомами и имеют схожие проявления ; первый вызван дефектной лизосомальной α-маннозидазой , а второй — дефектной лизосомальной β-маннозидазой . В обоих случаях дефект вызывает накопление олигосахаридов , богатых маннозой. [ 2 ] в нервной ткани и тканях органов . [ 3 ] Известно, что альфа- и бета-маннозидоз являются результатом аутосомно- рецессивных генетических мутаций. [ 4 ]
Альфа-маннозидоз
[ редактировать ]Альфа-маннозидоз — наследственное лизосомальное заболевание накопления , вызывающее потерю слуха , умственную отсталость , аномалии лица и скелета, а также иммунологические дефициты. К основным характеристикам относятся скелетные аномалии, нарушения слуха , постепенное ухудшение психических функций и речи, частые периоды психоза . Иммунная недостаточность проявляется рецидивирующими инфекциями, особенно в первом десятилетии жизни. Аутосомно-рецессивные мутации гена MAN2B1, расположенного на 19-й хромосоме (19 p13.2-q12), являются причиной альфа-маннозидоза . Активность кислой альфа-маннозидазы в лейкоцитах или других ядросодержащих клетках используется для постановки диагноза, который затем можно подтвердить с помощью генетического тестирования . [ 5 ]
Бета-маннозидоз
[ редактировать ]Бета-маннозидоз — крайне редкое метаболическое заболевание накопления, обусловленное дефицитом фермента бета-маннозидазы , участвующего в расщеплении гликопротеинов . Клинические проявления могут варьироваться от легких до тяжелых и включают такие симптомы, как потеря слуха , умственная отсталость , гиперактивность, поведенческие проблемы, а также рецидивирующие респираторные и кожные инфекции. [ 6 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Маннозидоз Национальной медицинской библиотеки США в медицинских предметных рубриках (MeSH)
- ^ Эллисон, Дэвид; С любовью, Сет; Чимелли, Лейла; Хардинг, Брайан Н.; Лоу, Джеймс С.; Винтерс, Гарри В.; Бранднер, Себастьян; Йонг, Уильям Х. (2013), «Лизосомальные и пероксисомальные расстройства» , Neuropathology , Elsevier, стр. 479–498, doi : 10.1016/b978-0-7234-3515-0.00023-4 , ISBN 978-0-7234-3515-0 , получено 3 января 2024 г.
- ^ Мин, Джеффри Э.; Грэм, Джон М. (1 января 2014 г.), Салливан, Кэтлин Э.; Стим, Э. Ричард (ред.), «Глава 12 - Генетические синдромы с признаками иммунодефицита» , Иммунные дефициты Стима , Амстердам: Academic Press, стр. 281–324, doi : 10.1016/b978-0-12-405546- 9.00012-1 , ISBN 978-0-12-405546-9 , получено 3 января 2024 г.
- ^ Руда, День; Нильссен, Ойвинд (декабрь 2008 г.). «Альфа-маннозидоз» . Сиротский журнал редких заболеваний . 3 (1): 21. дои : 10.1186/1750-1172-3-21 . ISSN 1750-1172 . ПМК 2515294 . ПМИД 18651971 .
- ^ Руда, День; Нильссен, Ойвинд (2008). «Альфа-маннозидоз» . Сиротский журнал редких заболеваний . 3 (1): 21. дои : 10.1186/1750-1172-3-21 . ISSN 1750-1172 . ПМК 2515294 . ПМИД 18651971 .
- ^ Сафка Брозкова, Дана; Варга, Лукас; Ухрова Месаросова, Анна; Слободова, Зузана; Скопкова, Мартина; Солтысова, Андреа; Фичек, Андрей; Енцик, Ян; Ластувкова Яна; Гасперикова, Даниэла; Зееман, Павел (2020). «Вариант c.2158-2A>G в MANBA является важной и частой причиной наследственной потери слуха и бета-маннозидоза среди чешских и словацких цыган – свидетельство нового этнического варианта» . Сиротский журнал редких заболеваний . 15 (1): 222. дои : 10.1186/s13023-020-01508-3 . ISSN 1750-1172 . ПМЦ 7448337 . ПМИД 32847582 .