Альфа-маннозидоз
Эта статья нуждается в дополнительных цитатах для проверки . ( июль 2008 г. ) |
| Альфа-маннозидоз | |
|---|---|
 | |
| Альфа-маннозидоз имеет аутосомно-рецессивный тип наследования. Рисунок 1 | |
| Специальность | Эндокринология |
Альфа-маннозидоз – лизосомальное заболевание накопления . [ 1 ] впервые описан шведским врачом Окерманом в 1967 году. [ 2 ] Известно, что у человека оно вызвано аутосомно- рецессивной генетической мутацией в гене MAN2B1, расположенном на хромосоме 19, влияющей на выработку фермента альфа-D-маннозидазы, что приводит к его дефициту. [ 2 ] [ 3 ] [ 4 ] Следовательно, если оба родителя являются носителями, при каждой беременности существует 25% вероятность того, что дефектный ген от обоих родителей будет унаследован и у ребенка разовьется заболевание. Существует два шанса из трех, что здоровые братья и сестры будут носителями (рис. 1). [ 4 ] У домашнего скота альфа-маннозидоз вызывается хроническим отравлением сваинсонином из рябины . [ 5 ]
Симптомы
[ редактировать ]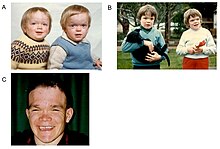
Альфа-маннозидоз — это мультисистемное прогрессирующее заболевание, продолжающееся в течение всей жизни, с ухудшением нервно-мышечной и скелетной функций в течение десятилетий. [ 2 ] Время появления симптомов коррелирует с тяжестью заболевания. Начало наиболее тяжелой формы заболевания происходит в первые месяцы жизни и включает скелетные аномалии и умственную отсталость , с быстрым прогрессированием, приводящим к смерти от первичного поражения центральной нервной системы или миопатии. [ 2 ] Однако у большинства новорожденных с лизосомальными нарушениями накопления симптомы заболевания протекают бессимптомно и лишь в редких случаях они серьезно поражаются. [ 1 ] [ 6 ] Это задерживает диагностику, особенно потому, что более легкие формы заболевания сопровождаются лишь легкой или умеренной умственной отсталостью, которая постепенно прогрессирует в детстве или подростковом возрасте. [ 7 ]
Первое десятилетие жизни характеризуется развитием нарушений слуха, задержкой психомоторного развития, рецидивирующими инфекциями, особенно верхних дыхательных путей, легочными инфекциями и острыми/серозными инфекциями среднего отита. [ 8 ] Могут возникнуть значительные изменения ряда черт лица, таких как выступающий лоб, уплощенная переносица, маленький нос, широкий рот и широко расставленные зубы. [ 2 ] Мышечная слабость или аномалии позвоночника могут возникнуть из-за накопления запасов веществ в мышцах. [ 2 ]
Патофизиология
[ редактировать ]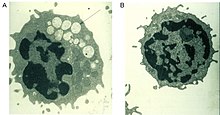
Дефектный фермент альфа-маннозидаза, который обычно помогает расщеплять сложные сахара, полученные из гликопротеинов в лизосомах , вызывает прогрессирующее лизосомальное накопление богатых маннозой олигосахаридов во всех тканях, что приводит к нарушению клеточной функции и апоптозу (рис. 2). [ 2 ] [ 9 ] Полное отсутствие функциональности этого фермента приводит к смерти в раннем детстве из-за ухудшения состояния центральной нервной системы . [ 9 ] Ферменты с низкой остаточной активностью приводят к более легкой форме заболевания с такими симптомами, как нарушение слуха, когнитивные нарушения, восприимчивость к бактериальным инфекциям и деформации скелета. Течение заболевания прогрессирующее. [ 2 ] [ 9 ]
В зависимости от тяжести заболевания альфа-маннозидоз подразделяют на три предполагаемых подтипа в зависимости от тяжести и возраста начала заболевания. [ 2 ]
- Тип 1: легкая форма, диагностируемая после десяти лет, с отсутствием скелетных аномалий, мышечных проблем (миопатии) и медленным прогрессированием.
- Тип 2: умеренная форма, диагностируемая в возрасте до десяти лет, с наличием скелетных аномалий, миопатии и медленным прогрессированием. Это наиболее распространенная форма
- Тип 3: Тяжелая форма, приводящая к ранней смерти из-за прогрессирующего поражения центральной нервной системы.
Однако, учитывая разнообразие зарегистрированных мутаций, а также широкий спектр и тяжесть симптомов, заболевание рассматривается клинически как континуум. [ 9 ] [ 8 ]
Диагностика
[ редактировать ]Альфа-маннозидоз является прогрессирующим заболеванием, и его наличие следует подозревать у пациентов с когнитивными нарушениями, изменениями скелета (например, опухшими суставами, искривлением позвоночника), потерей слуха и рецидивирующими инфекциями. Хотя дети с этим заболеванием часто рождаются на вид нормальными, с возрастом их состояние ухудшается. Альфа-маннозидоз может влиять на качество жизни пациента во многих отношениях, включая его способность жить независимо, общаться или находить работу. [ 2 ] [ 8 ]
Как правило, фенотипы пациентов с альфа-маннозидозом четко не различимы, что затрудняет прогнозирование клинического течения у отдельного пациента. [ 2 ] Пациенты могут обращаться к врачам, медсестрам или патронажным сестрам на разных стадиях прогрессирования и с разными симптомами , что затрудняет подозрение на диагноз альфа-маннозидоза. [ 2 ] Основные симптомы также могут быть схожи с симптомами других лизосомальных нарушений накопления, таких как мукополисахаридоз . [ 2 ]
Учитывая прогрессирующий характер заболевания, чем раньше будет поставлен правильный диагноз, тем лучше. [ 2 ] Это состояние часто диагностируется и лечится с использованием междисциплинарного подхода с участием педиатров, ортопедов, офтальмологов, отологов, неврологов, иммунологов, нейрохирургов и физиотерапевтов. [ 8 ]
Диагноз альфа-маннозидоза подозревается на основании выявления характерных признаков многосимптомной картины, тщательного клинического обследования, подробного анамнеза пациента и результатов диагностических тестов, описанных ниже:
А. Олигосахариды в моче
Предварительное исследование может быть проведено для измерения концентрации олигосахаридов, богатых маннозой, в моче. Повышенная экскреция с мочой олигосахаридов, богатых маннозой, позволяет предположить, но не является диагностическим признаком заболевания. [ 2 ]
B. Активность кислой альфа-маннозидазы
Диагноз подтверждается путем измерения остаточной активности альфа-маннозидазы в лейкоцитах или других ядросодержащих клетках с помощью флуорометрического анализа. [ 2 ] Это самый надежный метод диагностики, наряду с генетическим тестированием .
C. Генетическое тестирование
Идентификация мутаций, вызывающих заболевание, достигается с использованием ДНК клеток периферической крови путем полимеразной цепной реакции (ПЦР) амплификации всех 24 экзонов MAN2B1 с последующим секвенированием ДНК. [ 2 ]
Уход
[ редактировать ]Лекарства от врожденного альфа-маннозидоза не существует, и в целом подход к ведению носит превентивный характер с целью предотвращения возникающих осложнений. После полного медицинского обследования врачи должны сосредоточиться на известных осложнениях альфа-маннозидоза, таких как гидроцефалия , средний отит , потеря слуха, кариес зубов, суставные симптомы, кифосколиоз и психическое состояние. [ 2 ] Лечение часто ограничивается уменьшением или контролем симптомов заболевания с помощью, например, лекарств для контроля судорог, слуховых аппаратов для облегчения потери слуха и обычной физиотерапии для облегчения мышечных болей и слабости. [ 2 ] В некоторых случаях инвалидная коляска может быть уместна, если мышечные или спинальные нарушения обездвиживают пострадавшего.
Трансплантация гемопоэтических стволовых клеток (ТГСК) может быть вариантом лечения для некоторых пациентов, однако соотношение риска и пользы более благоприятно у более молодых пациентов, поэтому обеспечение ранней диагностики имеет решающее значение для того, чтобы этот вариант был жизнеспособным. [ 2 ] Обоснование состоит в том, что донорские клетки, продуцирующие ферменты, заселяют ткань хозяина и переносят фермент в близлежащие клетки-хозяева с дефицитом ферментов. [ 2 ] Несмотря на ранние сообщения об обратном, [ 10 ] [ 11 ] [ 2 ] Возможные преимущества ТГСК должны быть сопоставлены с общим риском заболеваемости и смертности, связанных с процедурой. Преимущества больше у молодых пациентов до того, как развились осложнения, а также осложнения, связанные с трансплантацией, более часты и тяжелы у пожилых пациентов.
Ферментозаместительная терапия (ФЗТ) является терапевтической альтернативой при ряде лизосомальных болезней накопления. [ 2 ] [ 8 ] Общий принцип ФЗТ заключается в том, что рекомбинантно полученная версия дефицитного фермента вводится в кровоток, откуда он интернализуется клетками и достигает лизосом посредством захвата, опосредованного маннозо-6-фосфатным рецептором, заменяя тем самым недостающий эндогенный фермент. . [ 8 ] ФЗТ с велманазой альфа одобрена для использования в Европейском Союзе и США. [ 12 ]
Прогноз
[ редактировать ]Долгосрочный прогноз состояния плохой. [ 2 ] Обычно нервно-мышечные и костные изменения прогрессируют медленно в течение десятилетий. Также могут присутствовать поведенческие проблемы или психические расстройства. [ 2 ] [ 8 ] Ожидаемая продолжительность жизни при альфа-маннозидозе сильно варьирует. Лица с ранним началом тяжелого заболевания часто не выживают после детства, тогда как люди с более легкими расстройствами могут дожить до взрослой жизни.
Самостоятельная жизнь будет затруднена, пациенты с альфа-маннозидозом могут оказаться в социальной изоляции, а на поздних стадиях заболевания они могут стать инвалидами-колясочниками, поскольку больше не смогут передвигаться без посторонней помощи. [ 2 ] Это, вероятно, окажет негативное влияние на качество жизни лиц, осуществляющих уход, и членов семьи. [ 2 ] [ 8 ]
Эпидемиология
[ редактировать ]Мировая заболеваемость альфа-маннозидозом точно неизвестна. Тем не менее, по оценкам ряда отчетов из разных стран, это заболевание встречается примерно у одного из миллионов младенцев, рожденных во всем мире. [ 9 ] Маннозидоз встречается у всех этнических групп Европы, Америки, Африки и Азии. [ 2 ]
Ссылки
[ редактировать ]- ^ Jump up to: а б Росес Д.П., Люльманн-Раух Р., Пэн Дж. и др. (2004). «Эффективность заместительной ферментной терапии у мышей с альфа-маннозидозом: доклиническое исследование на животных» . Хм. Мол. Жене . 13 (18): 1979–88. дои : 10.1093/hmg/ddh220 . ПМИД 15269179 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб Мальм Д., Нильссен О (2008). «Альфа-маннозидоз». Orphanet J Редкий дис. 3 (1): 21. дои : 10.1186/1750-1172-3-21 PMC 2515294. ПМИД 18651971
- ^ Готода Ю., Вакамацу Н., Каваи Х., Нисида Ю., Мацумото Т. (октябрь 1998 г.). «Миссенс- и нонсенс-мутации в лизосомальном гене альфа-маннозидазы (MANB) при тяжелых и легких формах альфа-маннозидоза» . Американский журнал генетики человека . 63 (4): 1015–24. дои : 10.1086/302048 . ПМЦ 1377481 . ПМИД 9758606 .
- ^ Jump up to: а б База данных мутаций альфа-маннозидоза. Университет Тромсё. Доступно по адресу: [1] .
- ^ Стегельмайер, БЛ; Джеймс, Л.Ф.; Пантер, Кентукки; Ральфс, Миннесота; Гарднер, доктор медицинских наук; Молинье, Р.Дж.; Пфистер, Дж. А. (февраль 1999 г.). «Патогенез и токсикокинетика отравлений жимолости (Astragalus и Oxytropis spp.) у домашнего скота» . Журнал природных токсинов . 8 (1). ПМИД 10091126 .
- ^ Альфа-маннозидоз. Информационный бюллетень Национальной организации по редким заболеваниям (NORD), 2015 г. https://rarediseases.org/rare-diseases/alpha-mannosidosis/
- ^ Руководство по пониманию маннозидоза. Общество мукополисахаридных заболеваний. http://www.mpssociety.org.uk/wp-content/uploads/2016/07/guide-alphamannosidosis-2013.pdf
- ^ Jump up to: а б с д и ж г час Боргвардт Л., Лунд А.М., Дали К.И. (2014). Альфа-маннозидоз – обзор генетических, клинических данных и вариантов лечения. Педиатр. Эндокринол. Откр. 12, Приложение 1:185-91.
- ^ Jump up to: а б с д и Бек М. Олсен К.Дж., Рэйт Дж.Э. и др. Естественная история альфа-маннозидоза: продольное исследование. Orphanet J Rare Dis 2013;8:88.
- ^ Уилл А и др. (1987). «Трансплантация костного мозга в лечении альфа-маннозидоза». Болезнь в детстве. 62 (10): 1044–1049. два : 10.1136/adc.62.10.1044
- ^ Гревал С.С., Шапиро Э.Г., Кривит В. и др. (2004). Эффективное лечение альфа-маннозидоза путем аллогенной трансплантации гемопоэтических стволовых клеток. Дж. Педиатр, 144:569-573.
- ^ «Ламзеде» . 17 сентября 2018 г.
Эта статья включает список общих ссылок , но в ней отсутствуют достаточные соответствующие встроенные цитаты . ( декабрь 2008 г. ) |
Дальнейшее чтение
[ редактировать ]- Запись GeneReviews/NCBI/NIH/UW об альфа-маннозидозе
- Записи OMIM об альфа-маннозидозе
- Альфа-маннозидоз типа 1 в НИЗ Отделе редких заболеваний
- Альфа-маннозидоз типа 2 в НИЗ Отделе редких заболеваний