Борилирование
Реакции борилирования C–H, катализируемые металлами, представляют собой органические реакции, катализируемые переходными металлами, которые производят борорганическое соединение посредством функционализации алифатических и ароматических связей C–H и, следовательно, являются полезными реакциями для активации связей углерод-водород . [ 1 ] В реакциях борилирования C–H, катализируемых металлами, используются переходные металлы для прямого преобразования связи C–H в связь C–B. Этот путь может быть выгодным по сравнению с традиционными реакциями борилирования за счет использования дешевого и обильного углеводородного исходного материала, ограничения предварительно функционализированных органических соединений, уменьшения токсичных побочных продуктов и оптимизации синтеза биологически важных молекул. [ 2 ] [ 3 ] Бороновые кислоты и эфиры бороновой кислоты представляют собой распространенные борильные группы, включаемые в органические молекулы посредством реакций борилирования. [ 4 ] Бороновые кислоты – трехвалентные борсодержащие органические соединения, имеющие один алкильный заместитель и две гидроксильные группы. Аналогичным образом, эфиры бороновой кислоты имеют один алкильный заместитель и две сложноэфирные группы. Бороновые кислоты и сложные эфиры классифицируются в зависимости от типа углеродной группы (R), непосредственно связанной с бором, например алкил-, алкенил-, алкинил- и арилбороновые сложные эфиры. Наиболее распространенный тип исходных материалов, которые включают сложные эфиры бороновой кислоты в органические соединения для реакций борилирования, катализируемых переходными металлами, имеет общую формулу (RO) 2 B-B(OR) 2 . Например, бис(пинаколато)дибор (B 2 Pin 2 ) и бис(катехолато)диборан (B 2 Cat 2 ) являются обычными источниками бора этой общей формулы. [ 5 ]
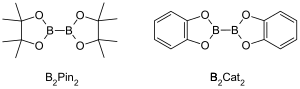
Атом бора боронового эфира или кислоты имеет вид sp. 2 гибридизуются с вакантной p-орбиталью, что позволяет этим группам действовать как кислоты Льюиса . Связь C–B бороновых кислот и сложных эфиров немного длиннее, чем типичные одинарные связи C–C, в диапазоне 1,55–1,59 Å. Удлиненная связь C–B относительно связи C–C приводит к тому, что энергия связи также немного меньше, чем у связей C–C (323 кДж/моль для C–B против 358 кДж/моль для C–C). [ 6 ] Связь углерод-водород имеет длину около 1,09 Å и энергию связи около 413 кДж/моль. Таким образом, связь C–B является полезным промежуточным соединением в качестве связи, которая заменяет обычно нереакционноспособную связь C–H.
Борорганические соединения – это органические соединения, содержащие связь углерод-бор. Борорганические соединения имеют широкое применение в химическом синтезе, поскольку связь C–B легко превращается в связь C–X (X = Br, Cl), C–O, C–N или C–C. Из-за универсальности связи C–B было разработано множество процессов для их включения в органические соединения. [ 7 ] Борорганические соединения традиционно синтезируются из реактивов Гриньяра посредством реакций гидроборирования или диборирования. [ 8 ] Борилирование обеспечивает альтернативу.
Реакции борилирования C – H, катализируемые металлами.
[ редактировать ]Алифатическое борилирование C – H
[ редактировать ]Как впервые описал Хартвиг, алканы можно селективно борилировать с высокой селективностью по первичной связи C–H, используя Cp*Rh(η 4 -C 6 Me 6 ) в качестве катализатора. [ 9 ] Примечательно, что селективность по первичной связи C–H является исключительной даже при наличии гетероатомов в углерод-водородной цепи. Катализируемое родием борилирование метильных связей С–Н происходит селективно, независимо от положения гетероатома. Борилирование происходит избирательно по наименее стерически затрудненной и наименее богатой электронами первичной связи C–H в ряде ацеталей , простых эфиров , аминов и алкилфторидов. [ 10 ] Кроме того, показано, что никакая реакция не протекает в отсутствие первичных связей C–H, например, когда циклогексан субстратом является .
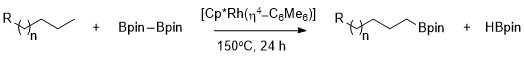
Селективная функционализация первичной алкановой связи происходит за счет образования кинетически и термодинамически выгодного первичного комплекса алкил-металл по сравнению с образованием вторичного комплекса алкил-металл. [ 11 ]

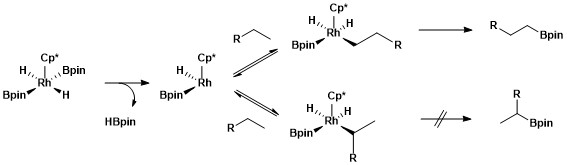
Большую стабильность первичных алкильных комплексов по сравнению с вторичными можно объяснить несколькими факторами. Во-первых, первичный алкильный комплекс стерически предпочтительнее вторичного алкильного комплекса. Во-вторых, на α-углероде комплекса металл-алкил часто присутствуют частичные отрицательные заряды, и первичный алкильный лиганд поддерживает частичный отрицательный заряд лучше, чем вторичный алкильный лиганд. Природа селективности алифатического борилирования C–H с использованием родиевых катализаторов была исследована с помощью механистического исследования, называемого водородно-дейтериевым обменом . Обмен H/D показал, что региоселективность всего процесса, показанного ниже, является результатом селективного расщепления первичных связей C–H по сравнению с вторичными и селективной функционализации первичного промежуточного металл-алкила по сравнению с промежуточным соединением вторичный металл-алкил. [ 12 ]
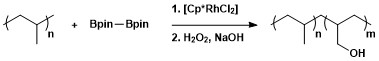
Синтетическая полезность алифатического борилирования C – H была применена для модификации полимеров посредством борилирования с последующим окислением с образованием полимеров с гидроксильными функциональными группами. [ 13 ]
Ароматическое борилирование C – H
[ редактировать ]Стерически направленное C–H борилирование аренов.
[ редактировать ]О первом примере каталитического C–H-борилирования неактивированного углеводорода (бензола) сообщили Смит и Айверсон с использованием Ir(Cp*)(H)(Bpin) в качестве катализатора. Однако эффективность этой системы была низкой: всего 3 оборота за 120 часов при 150 °С. [ 14 ] Многочисленные последующие разработки Хартвига и его коллег привели к созданию эффективных и практических условий борилирования аренов. Ароматическое борилирование C–H было разработано Джоном Ф. Хартвигом и Исиямой с использованием диборного реагента Bis(pinacolato)diboron, катализируемого 4,4'-ди-трет-бутилбипиридином (dtbpy) и [Ir(COD)(OMe)] 2 . [ 15 ] В данной каталитической системе борилирование ароматических связей C–H происходит с региоселективностью, контролируемой стерическими эффектами исходного арена. Селективность функционализации ароматических связей C–H определяется общим правилом, согласно которому реакция не протекает в орто- связь C–H, не имеющая орто- заместителя. заместителе, когда имеется [ 11 ] Когда присутствует только одна функциональная группа, борилирование происходит в мета- и пара -положении в статистических соотношениях 2:1 (мета:пара). Орто - изомер не обнаружен из-за стерических эффектов заместителя. [ 16 ]

Присоединение Bpin происходит только в одном положении для симметрично замещенных 1,2- и 1,4-замещенных аренов. Симметричные или несимметричные 1,3-замещенные арены также избирательно борилируются, поскольку стерически доступна только одна связь C–H.
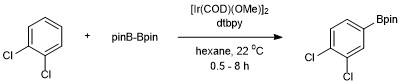
В этом отличие от электрофильного ароматического замещения , при котором региоселективность определяется электронными эффектами. [ 17 ]
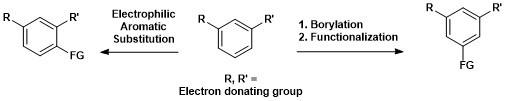
Синтетическая важность ароматического борилирования C–H показана ниже, где 1,3-дизамещенное ароматическое соединение может быть напрямую преобразовано в 1,3,5-органоборановое соединение и впоследствии функционализировано. [ 15 ]

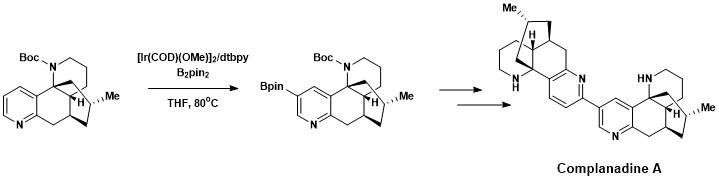
Функционализация ароматического C–H была успешно включена в общий синтез компладина А, Lycopodium алкалоида , который усиливает мРНК экспрессию фактора роста нервов (NGF) и продукцию NGF в глиальных клетках человека . Натуральные продукты, способствующие росту новых нейронных сетей, представляют интерес для лечения таких заболеваний, как болезнь Альцгеймера . [ 18 ] Компланадин А был успешно синтезирован с использованием комбинации прямого ароматического борилирования C–H, разработанной Хартвигом и Ишьямой, с последующим перекрестным сочетанием Сузуки-Мияуры и последующим расщеплением Boc-защитной группы .
C–H борилирование гетероаренов
[ редактировать ]Гетероарены также могут подвергаться борилированию в условиях, катализируемых иридием, однако сайт-селективность в этом случае контролируется электронными эффектами , когда фураны , пирролы и тиофены вступают в реакцию по альфа-связи C–H с гетероатомом. В этом случае предполагается, что селективность осуществляется через альфа-связь CH–H к гетероатому, поскольку это наиболее кислая связь C–H и, следовательно, наиболее реакционноспособная. [ 11 ]

Направленное орто- C–H-борилирование
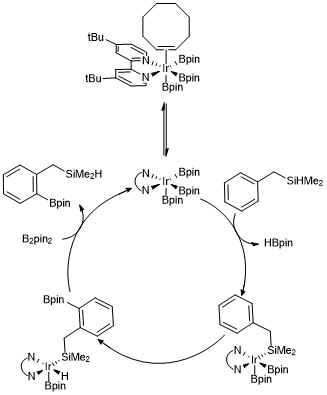
[ редактировать ]Используя ту же каталитическую систему, можно использовать направляющие группы для достижения региоселективности без заместителей в качестве стерических медиаторов. Например, Бебель и Хартвиг сообщили о методе проведения орто -борилирования, при котором диметилгидросилильная направляющая группа арена подвергается катализируемому иридием борилированию по связи C–H орто по отношению к силановой направляющей группе. [ 19 ] Селективность по орто- положению в случае использования гидросилильных направляющих групп объясняется обратимым присоединением связи Si-H к металлическому центру, что приводит к преимущественному разрыву орто- связи C-H по гидросилильному заместителю. Несколько других стратегий достижения орто -борилирования аренов были разработаны с использованием различных направляющих групп. [ 20 ] [ 21 ] [ 22 ]

Детали механизма C–H-борлиирования аренов.
[ редактировать ]Комплекс трисборила иридия был предложен для облегчения механизма каждой из этих реакций, которые приводят к борилированию C – H аренов и гетероаренов. Кинетические исследования и исследования изотопного мечения показали, что триборильный комплекс Ir(III) реагирует с ареном в каталитическом процессе. [ 23 ] Ниже показан вариант каталитического цикла орто- борилирования гидросилановых соединений. Кинетические данные показывают, что наблюдаемый трисборильный комплекс, координированный с циклооктеном, быстро и обратимо диссоциирует циклооктен с образованием 16-электронного трисборильного комплекса. В случае использования бензилдиметилсилана в качестве направляющей группы предполагается, что бензилдиметилсилан реагирует с трисборилиридиевым катализатором путем обратимого присоединения связи Si-H к металлическому центру с последующей селективной орто активацией связи -C–H путем окислительного присоединения и восстановительного присоединения. устранение . [ 24 ]
Метаселективное борилирование : Метаселективное борилирование C–H является важным синтетическим преобразованием, которое было обнаружено в 2002 году Смитом III из Университета штата Мичиган, США. Однако это метаборилирование носило полностью стерический характер и ограничивалось только 1,3-дизамещенными бензолами. Примерно 12 лет спустя доктор Чаттопадхьяй и его команда из Центра биомедицинских исследований, УП, Индия, открыли элегантную технологию метаселективной активации и борилирования связи C–H. Команда показала, что, используя тот же субстрат, можно переключить позиционную селективность другого, просто изменив лиганд. Происхождение метаселективности определялось двумя параметрами, такими как: 1) электростатическое взаимодействие, 2) вторичное взаимодействие BN. [ 25 ]
В то же время группа из Японии, доктор Канаи, сообщила об удивительной концепции метаселективного борилирования, основанной на вторичном взаимодействии. Этот метод охватывает борилирование различных карбонильных соединений. [ 26 ]
Реакции восстановления с борорганическими соединениями
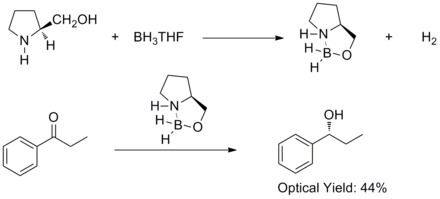
[ редактировать ]В 1981 году Хирао и его коллеги обнаружили, что асимметричное восстановление прохиральных ароматических кетонов хиральными аминоспиртами и бораном дает соответствующие вторичные спирты с 60% ее . Они обнаружили, что хиральные аминоспирты реагируют с бораном с образованием комплексов алоксиламин-боран. Предполагается, что комплексы содержат относительно жесткую пятичленную кольцевую систему, что делает их термически и гидролитически стабильными и растворимыми в широком спектре протонных и апротонных растворителей. [ 27 ]
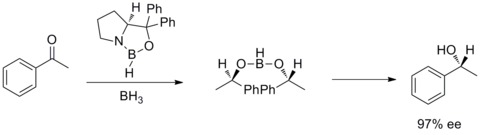
В 1987 году Элиас Джеймс Кори и его коллеги обнаружили возможность образования оксазаборолидинов из борана и хиральных аминоспиртов. Было обнаружено, что оксазаборолидины катализируют быстрое и высокоэнантиоселективное восстановление прохиральных кетонов в присутствии BH3THF. Это энантиоселективное восстановление ахиральных кетонов каталитическим оксазаборолидином называется восстановлением Кори-Бакши-Шибаты или восстановлением CBS. [ 28 ] [ 29 ]
В 1977 году М.М. Мидланд и его коллеги сообщили об удивительном наблюдении, что B-3-альфа-пинанил-9-борабицикло[3,3,1]нонан легко получается гидроборированием (+)-альфа-пинена с 9-боробицикло. [3,3,1]нонан быстро восстанавливает бензальдегид-альфа-d до (S)-(+)-бензил-альфа-d спирта с существенно количественная асимметричная индукция. [ 30 ]
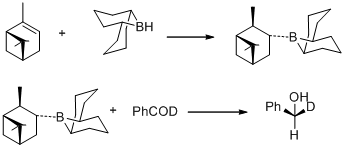
В том же году М.М. Мидланд обнаружил B-3-альфа-пинанил-9-BBN в качестве восстановителя, который можно легко получить путем реакции (+)-альфа-пинена с 9-BBN. Новый восстановитель был позже коммерциализирован компанией Aldrich Co. под названием Alpine Borane , а асимметричное восстановление карбонильных групп с помощью любого энантиомера альпийского борана известно как восстановление Midland Alpine-Boran. [ 31 ]
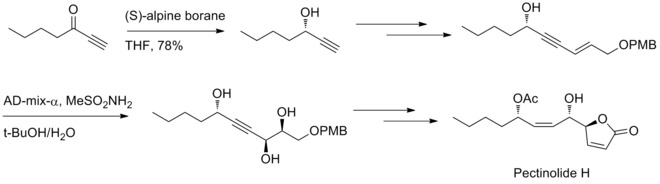
В 2012 году УРИ Венкатешварлу и его коллеги сообщили о стереоселективном методе синтеза пектинолида H. Восстановление Мидленда и реакция дигидроксилирования Шарплесса участвуют в создании трех хиральных центров при C-4', C-5 и C-1'. [ 32 ]
Реакции сочетания с борорганическими соединениями
[ редактировать ]В 1993 г. Н. А. Петасис и И. Акрлтопулу сообщили об эффективном синтезе аллильных аминов с помощью модифицированной реакции Манниха . В этой модифицированной реакции Манниха они обнаружили, что винилбороновые кислоты могут участвовать в качестве нуклеофилов, образуя геометрически чистые аллиламины. Эта модифицированная реакция Манниха была известна как реакция Петасиса бороновой кислоты-Манниха. [ 33 ] [ 34 ]
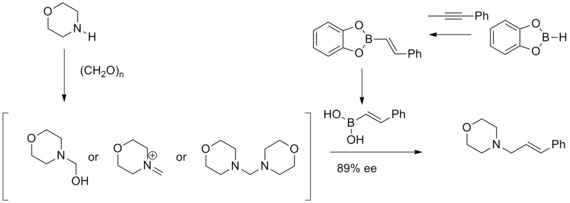
Асимметричная аллилация Руша
[ редактировать ]В 1978 году Р.В. Хоффманн и Т. Герольд сообщили об энантиоселективном синтезе вторичных гомоаллиловых спиртов посредством хиральных нерацемических аллилбороновых эфиров . Гомоаллильные спирты образуются с отличным выходом и умеренной энантиоселективностью. [ 35 ]
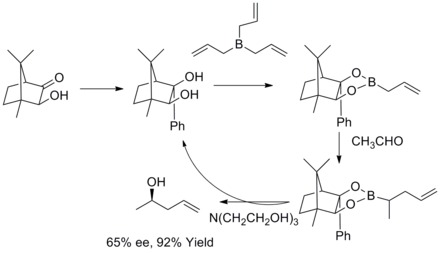
В 1985 году WR Roush и его коллеги обнаружили, что модифицированные тартратом аллильные боронаты предлагают простой и очень привлекательный подход к контролю лицевой селективности в реакциях с хиральными и ахиральными альдегидами. В последующие годы WR Roush и его коллеги расширили эту стратегию до синтеза бут-2-ен-1,4-диолов и антидиолов . Этот тип реакции известен как асимметричное аллилирование Руша. [ 36 ] [ 37 ] [ 38 ] [ 39 ]
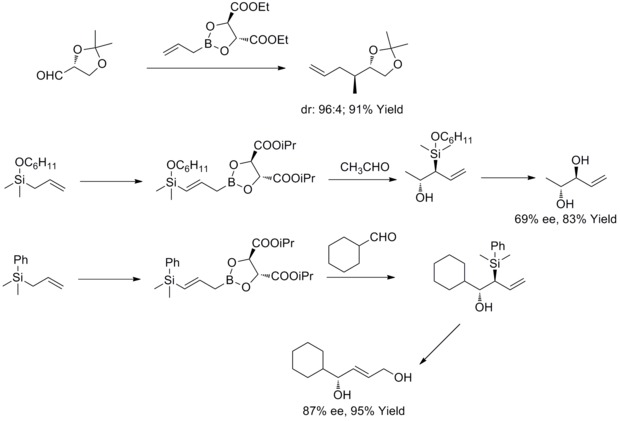
В 2011 году Р. А. Фернандес и П. Каттангуру завершили улучшенный общий синтез диастереомеров (8S, 11R, 12R)- и (8R, 11R, 12R)-топсентолида B2 в восемь этапов. В статье диастереоселективная реакция аллилирования Рауша использовалась в качестве ключевой реакции в общем синтезе для введения двух хиральных промежуточных продуктов. А затем авторы синтезировали два диастереомера через эти два хиральных промежуточных продукта. [ 40 ]

В 1979 г. Н. Мияура и А. Судзуки сообщили о синтезе арилированных (Е)-алкенов с высоким выходом из арилгалогенидов с алкил-1-енилборанами, катализируемом тетракис( трифенилфосфин )палладием и основаниями. Затем А. Судзуки с сотрудниками распространили этот вид реакции на другие борорганические соединения и другие алкенил, арил , алкилгалогениды и трифлаты . Катализируемая палладием реакция кросс-сочетания борорганических соединений и этих органических галогенидов с образованием углерод-углеродных связей известна как кросс-сочетание Сузуки-Мияуры . [ 41 ] [ 42 ]
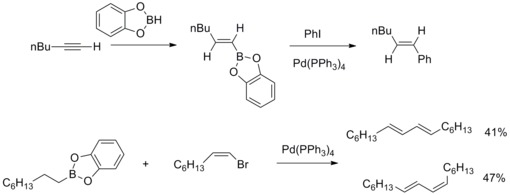
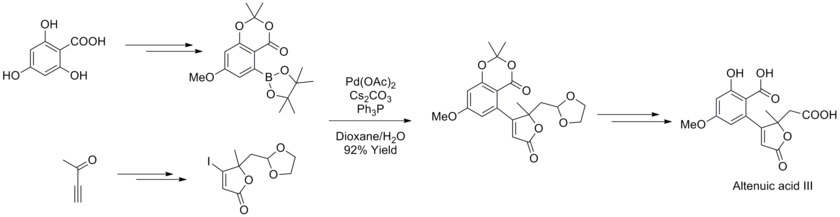
В 2013 году Иоахим Подлех и его коллеги определили структуру микотоксина Alternaria альтенуиновой кислоты III с помощью ЯМР-спектроскопического анализа и завершили его полный синтез. В синтетической стратегии использовалась реакция кросс-сочетания Сузуки-Мияуры с высокофункционализированным боронатом и бутенолидами для синтеза предшественника природного продукта с высоким выходом. [ 43 ]
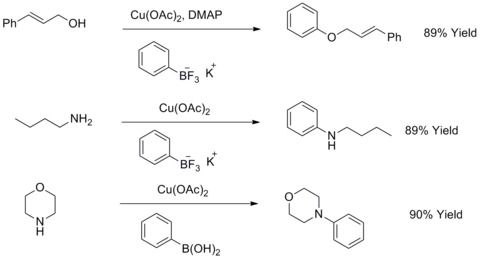
Синтез модифицированного биарилового эфира Ульмана и биариламина
[ редактировать ]В 1904 году Фриц Ульман обнаружил, что медный порошок может значительно улучшить реакцию арилгалогенидов с фенолами с образованием биариловых эфиров. Эта реакция известна как конденсация Ульмана . В 1906 И. Гольдберг распространил эту реакцию на синтез ариламина путем взаимодействия арилгалогенидов с амидом в присутствии карбоната калия и CuI. Эта реакция известна как модифицированная конденсация Ульмана по Гольдбергу. [ 44 ] В 2003 году Р. А. Бэти и Т. Д. Куах модифицировали реакции такого типа, используя соли органотрифторборатов калия для взаимодействия с алифатическими спиртами, алифатическими аминами или анилинами с целью синтеза ариловых эфиров или ариламинов. [ 45 ] [ 46 ]
См. также
[ редактировать ]- Борорганическая химия
- Реакции органоборатов и боранов
- Сокращение Кори-Ицуно
- Восстановление борана в Мидленд-Альпийских Альпах
- Реакция Петасиса
- Реакция Сузуки
Ссылки
[ редактировать ]- ^ Хартвиг, Джон Ф. (2012). «Борилирование и силилирование связей C–H: платформа для разнообразной функционализации связей C–H». Отчеты о химических исследованиях . 45 (6): 864–873. дои : 10.1021/ar200206a . ISSN 0001-4842 . ПМИД 22075137 .
- ^ Чо, JY; Це, МК; Холмс, Д.; Малечка, Р.Э. младший; Смит, MR (2001). «Удивительно селективные иридиевые катализаторы для создания ароматических связей CH» . Наука . 295 (5553): 305–8. дои : 10.1126/science.1067074 . ПМИД 11719693 . S2CID 21096755 .
- ^ Исияма, Т.; Нобута, Ю.; Хартвиг, Дж. Ф.; Мияура, Н. Хим. Коммун. 2003 , 2924.
- ^ Браун, ХК; Крамер, Г.В.; Леви, AB; Мидленд, ММ. Органический синтез с помощью боранов ; Wiley-Interscience: Нью-Йорк, 1975; Том. 1.
- ^ Брауншвейг, Х.; Гетлейн, Ф. (2011). «Синтез диборанов (4), катализируемый переходными металлами». Angewandte Chemie, международное издание . 50 (52): 12613–12616. дои : 10.1002/anie.201104854 . ПМИД 22057739 .
- ^ Холл, Д.Г. (2011) Структура, свойства и получение производных бороновой кислоты, в Бороновые кислоты: получение и применение в органическом синтезе, медицине и материалах (том 1 и 2), второе издание (под ред. Д.Г. Холла), Wiley-VCH Verlag GmbH & Co. KGaA, Вайнхайм, Германия. два : 10.1002/9783527639328.ch1
- ^ Мхалид, Ибрагим А.И.; Барнард, Джонатан Х.; Мардер, Тодд Б.; Мерфи, Жаклин М.; Хартвиг, Джон Ф. (2010). «Активация C – H для создания облигаций C – B». Химические обзоры . 110 (2): 890–931. дои : 10.1021/cr900206p . ПМИД 20028025 .
- ^ Уэйд, Л.Г., Органическая химия . Река Аппер-Седл: Pearson Education, Inc., 2010 .
- ^ Чен, Х.; Шлехт, С.; Семпл, ТК; Хартвиг, Дж. Ф. (2000). «Термическая, каталитическая, региоспецифическая функционализация алканов». Наука . 287 (5460): 1995–1997. Бибкод : 2000Sci...287.1995C . дои : 10.1126/science.287.5460.1995 . ПМИД 10720320 .
- ^ Лоуренс, доктор юридических наук; Такахаши, М.; Бэ, К.; Хартвиг, Дж. Ф. (2004). «Региоспецифическая функционализация метил-СН-связей алкильных групп в реагентах с гетероатомной функциональностью». Дж. Ам. хим. Соц . 126 (47): 15334–15335. дои : 10.1021/ja044933x . ПМИД 15563132 .
- ^ Jump up to: а б с Хартвиг, Дж. Ф. (2011). «Региоселективность борилирования алканов и аренов». хим. Соц. Преподобный . 40 (4): 1992–2002. дои : 10.1039/C0CS00156B . ПМИД 21336364 .
- ^ Вэй, CS; Хименес-Ойос, Калифорния; Видео, МФ; Хартвиг, Дж. Ф.; Холл, МБ (2010). «Происхождение селективности борилирования первичных по сравнению с вторичными связями CH, катализируемого комплексами Cp *-родия». Дж. Ам. хим. Соц . 132 (9): 3078–91. дои : 10.1021/ja909453g . ПМИД 20121104 .
- ^ Кондо, Ю.; Гарсия-Куадрадо, Д.; Хартвиг, Дж. Ф.; Боэн, Северная Каролина; Вагнер, Нидерланды; Хиллмайер, Массачусетс (2002). «Родий-катализируемая региоспецифическая функционализация полиолефинов в расплаве». Дж. Ам. хим. Соц . 124 (7): 1164–5. дои : 10.1021/ja016763j . ПМИД 11841273 .
- ^ Айверсон, Карл Н.; Смит, Милтон Р. (6 августа 1999 г.). «Стехиометрическое и каталитическое образование связи BC из неактивированных углеводородов и боранов». Журнал Американского химического общества . 121 (33): 7696–7697. дои : 10.1021/ja991258w .
- ^ Jump up to: а б Хартвиг, Дж. Ф. (2012). «Борилирование и силилирование связей CH: платформа для разнообразной функционализации связей CH». Отчеты о химических исследованиях . 45 (6): 864–873. дои : 10.1021/ar200206a . ПМИД 22075137 .
- ^ Исияма, Т.; Такаги, Дж.; Исида, К.; Мияура, Н.; Анастази, Н.; Хартвиг, Дж. Ф. (2002). «Мягкое борилирование аренов, катализируемое иридием. Высокие числа оборотов, реакции при комнатной температуре и выделение потенциального промежуточного продукта». Дж. Ам. хим. Соц . 124 (3): 390–391. дои : 10.1021/ja0173019 . ПМИД 11792205 .
- ^ Лиски, К. Иридий-катализируемое борилирование ароматических и алифатических связей C–H: методология и механизм. Диссертация, Университет Иллинойса. Урбанан-Шампейн. 2013.
- ^ Фишер, Д.Ф.; Сарпонг, Р. (2010). «Полный синтез (+)-компланадина А с использованием иридий-катализируемой функционализации пиридина C-H» . Дж. Ам. хим. Соц . 132 (17): 5926–5927. дои : 10.1021/ja101893b . ПМК 2867450 . ПМИД 20387895 .
- ^ Бебель, Т.А.; Хартвиг, Дж. Ф. (2008). «Силил-направленное иридий-катализируемое дорто-борилирование аренов. Одно-поторто-борилирование фенолов, ариламинов и алкиларенов». Дж. Ам. хим. Соц . 130 (24): 7534–5. дои : 10.1021/ja8015878 . ПМИД 18494474 .
- ^ Исияма, Т.; Мияура, Н.; Ису, Х.; Кикучи, Т. (2010). «Орто-C – H борилирование эфиров бензойной кислоты бис (пинаколато) дибором, катализируемое иридий-фосфиновыми комплексами». хим. Коммун . 46 (1): 159–61. дои : 10.1039/b910298a . hdl : 2115/44631 . ПМИД 20024326 .
- ^ Каваморита, С.; Омия, Х.; Хара, К.; Фукуока, А.; Савамура, М. (2009). «Направленное ортоборилирование функционализированных аренов, катализируемое компактной системой фосфин-иридий на кремнеземе». Дж. Ам. хим. Соц . 131 (14): 5058–9. дои : 10.1021/ja9008419 . ПМИД 19351202 .
- ^ Роуз, А.; Стивен, Б.; Лопес-Родрикес, Р.; Альварес, Э.; Фернандес, Р.; Лассалетта, Дж. М. Энжью. хим. Они. Пшеница. 2011 г.; 50, 1.
- ^ Боллер, ТМ; Мерфи, Дж. М.; Хапке, М.; Шияма, Т.; Мияура, Н.; Хартвиг, Дж. Ф. Дж. Ам. Ткань. Соц. 2005;, 127, 14263.
- ^ Бебель, Т.А.; Хартвиг, Дж. Ф. (2008). «Силил-направленное иридий-катализируемое дорто-борилирование аренов. Одно-поторто-борилирование фенолов, ариламинов и алкиларенов». Дж. Ам. хим. Соц . 130 (24): 7534–7535. дои : 10.1021/ja8015878 . ПМИД 18494474 .
- ^ Бишт, Р.; Чаттопадхай, Б. (2016). «Формальное ИК-катализируемое лигандом орто- и метаборилирование ароматических альдегидов посредством иминов, генерируемых in Situ». Дж. Ам. хим. Соц . 138 (1): 84–7. дои : 10.1021/jacs.5b11683 . ПМИД 26692251 .
- ^ Канаи; и др. (2015). «Метаселективное борилирование C – H, направляемое вторичным взаимодействием между лигандом и субстратом». Нат. Хим . 7 (9): 712–7. Бибкод : 2015НатЧ...7..712К . дои : 10.1038/nchem.2322 . ПМИД 26291942 .
- ^ Хирао, Акира; Ицуно, Шиничи; Накахама, Сейичи; Ямадзаки, Нобору (1981). «Асимметричное восстановление ароматических кетонов хиральными алкоксиаминоборановыми комплексами». Журнал Химического общества, Chemical Communications (7): 315. doi : 10.1039/c39810000315 .
- ^ Кори, Э.Дж.; Бакши, Раман К.; Сибата, Сайзо (сентябрь 1987 г.). «Высокоэнантиоселективное борановое восстановление кетонов, катализируемое хиральными оксазаборолидинами. Механизм и синтетические последствия». Журнал Американского химического общества . 109 (18): 5551–5553. дои : 10.1021/ja00252a056 .
- ^ Кори, Э.Дж.; Бакши, Раман К.; Сибата, Сайзо; Чен, Чунг Пин; Сингх, Винод К. (декабрь 1987 г.). «Стабильный и легко приготовляемый катализатор энантиоселективного восстановления кетонов. Применение в многостадийном синтезе». Журнал Американского химического общества . 109 (25): 7925–7926. дои : 10.1021/ja00259a075 .
- ^ Мидленд, М.Марк; Трамонтано, Альфонсо; Здерич, Стивен А. (июль 1977 г.). «Легкая реакция B-алкил-9-борабицикло[3.3.1]нонанов с бензальдегидом». Журнал металлоорганической химии . 134 (1): С17–С19. дои : 10.1016/S0022-328X(00)93625-8 .
- ^ Мидленд, М. Марк; Трамонтано, Альфонсо; Здерич, Стивен А. (июнь 1977 г.). «Получение оптически активного бензил-альфа-d-спирта восстановлением B-3-альфа-пинанил-9-борабицикло[3.3.1]нонана. Новый высокоэффективный хиральный восстановитель». Журнал Американского химического общества . 99 (15): 5211–5213. дои : 10.1021/ja00457a068 .
- ^ Рамеш, Д.; Шехар, В.; Чантибабу, Д.; Раджарам, С.; Рамулу, У.; Венкатешварлу, Ю. (март 2012 г.). «Первый стереоселективный полный синтез пектинолида H». Буквы тетраэдра . 53 (10): 1258–1260. дои : 10.1016/j.tetlet.2011.12.122 .
- ^ Петасис, Никос А.; Акритопулу, Ирини (январь 1993 г.). «Реакция Манниха с бороновой кислотой: новый метод синтеза геометрически чистых аллиламинов». Буквы тетраэдра . 34 (4): 583–586. дои : 10.1016/S0040-4039(00)61625-8 .
- ^ Ю, Тао; Ли, Хуэй; У, Синьян; Ян, июнь (2012). «Прогресс в реакции Петасиса» . Китайский журнал органической химии . 32 (10): 1836. doi : 10.6023/cjoc1202092 .
- ^ Герольд, Томас; Хоффманн, Рейнхард В. (октябрь 1978 г.). «Энантиоселективный синтез гомоаллиловых спиртов с помощью хиральных аллилбороновых эфиров». Angewandte Chemie International Edition на английском языке . 17 (10): 768–769. дои : 10.1002/anie.197807682 .
- ^ Руш, Уильям Р.; Уолтс, Алан Э.; Хунг, Ли К. (декабрь 1985 г.). «Реакции диастерео- и энантиоселективного присоединения альдегидов сложных эфиров 2-аллил-1,3,2-диоксаборолан-4,5-дикарбоновых кислот, полезного класса аллилборонатов, модифицированных эфиром тартрата». Журнал Американского химического общества . 107 (26): 8186–8190. дои : 10.1021/ja00312a062 .
- ^ Руш, Уильям Р.; Андо, Каори; Пауэрс, Дэниел Б.; Халтерман, Рональд Л.; Палковиц, Алан Д. (январь 1988 г.). «Энантиоселективный синтез с использованием модифицированных диизопропилтартратом (E)- и (Z)-кротилборонатов: реакции с ахиральными альдегидами». Буквы тетраэдра . 29 (44): 5579–5582. дои : 10.1016/S0040-4039(00)80816-3 .
- ^ Руш, Уильям Р.; Гровер, Пол Т. (январь 1990 г.). «Диизопропилтартрат (E)-γ-(диметилфенилсилил)аллилборонат, эквивалент хирального аллильного спирта β-карбаниона для энантиоселективного синтеза 2-бутен-1,4-диолов из альдегидов». Буквы тетраэдра . 31 (52): 7567–7570. дои : 10.1016/S0040-4039(00)97300-3 .
- ^ Руш, Уильям Р.; Говер, Пол Т.; Линь, Сяофа (январь 1990 г.). «Диизопропилтартрат, модифицированный (E)-γ-[(циклогексилокси)диметилсилил-аллилборонат, хиральный реагент для стереоселективного синтеза анти-1,2-диолов посредством формального α-гидроксиаллилирования альдегидов». Буквы тетраэдра . 31 (52): 7563–7566. дои : 10.1016/S0040-4039(00)97299-X .
- ^ Фернандес, Родни А.; Каттангуру, Пуллайя (ноябрь 2011 г.). «Полный синтез диастереомеров (8S,11R,12R)- и (8R,11R,12R)-топсентолида В2 и определение абсолютной конфигурации». Тетраэдр: Асимметрия . 22 (20–22): 1930–1935. дои : 10.1016/j.tetasy.2011.10.020 .
- ^ Мияура, Норио; Сузуки, Акира (1979). «Стереоселективный синтез арилированных (Е)-алкенов реакцией алк-1-енилборанов с арилгалогенидами в присутствии палладиевого катализатора». Журнал Химического общества, Chemical Communications (19): 866. doi : 10.1039/C39790000866 .
- ^ Мияура, Норио; Ямада, Кинджи; Сузуки, Акира (январь 1979 г.). «Новое стереоспецифическое кросс-сочетание катализируемой палладием реакции 1-алкенилборанов с 1-алкенил- или 1-алкинилгалогенидами» (PDF) . Буквы тетраэдра . 20 (36): 3437–3440. дои : 10.1016/S0040-4039(01)95429-2 . hdl : 2115/44006 .
- ^ Немечек, Грегор; Томас, Роберт; Гёсманн, Гельмут; Фельдманн, Клаус; Подлех, Иоахим (октябрь 2013 г.). «Выяснение структуры и общий синтез альтеновой кислоты III и исследования полного синтеза альтеновой кислоты II». Европейский журнал органической химии . 2013 (28): 6420–6432. дои : 10.1002/ejoc.201300879 .
- ^ Курти, Ласло; Чако, Барбара (2007). Стратегическое применение названных реакций в органическом синтезе: предпосылки и детальные механизмы; 250 названных реакций (Пбк. ред., [Начдр.]. ред.). Амстердам [ua]: Elsevier Academic Press. стр. 464–465 . ISBN 978-0-12-429785-2 .
- ^ Куах, Тан Д.; Бэти, Роберт А. (апрель 2003 г.). «Синтез эфира, катализируемый медью (II), из алифатических спиртов и солей органотрифторбората калия». Органические письма . 5 (8): 1381–1384. дои : 10.1021/ol034454n . ПМИД 12688764 .
- ^ Куах, Тан Д.; Бэти, Роберт А. (1 ноября 2003 г.). «Образование связи C-N, не содержащей лигандов и оснований, катализируемое медью (II): реакции кросс-сочетания борорганических соединений с алифатическими аминами и анилинами». Органические письма . 5 (23): 4397–4400. дои : 10.1021/ol035681s . ПМИД 14602009 .