Присоединение соседа
В биоинформатике объединение соседей — это восходящий (агломеративный) метод кластеризации для создания филогенетических деревьев , созданный Наруей Сайто и Масатоши Неи в 1987 году. [1] Обычно алгоритм основан на ДНК или белков данных о последовательностях и требует знания расстояния между каждой парой таксонов (например, видов или последовательностей) для создания филогенетического дерева. [2]
Алгоритм
[ редактировать ]
Соединение соседей принимает в качестве входных данных матрицу расстояний , которая определяет расстояние между каждой парой таксонов .Алгоритм начинается с полностью неразрешенного дерева, топология которого соответствует топологии звездообразной сети , и повторяет следующие шаги, пока дерево не будет полностью разрешено и не станут известны длины всех ветвей:
- На основе текущей матрицы расстояний вычислите матрицу (определено ниже).
- Найдите пару различных таксонов i и j (т.е. с ) для чего самый маленький. Создайте новый узел, который объединяет таксоны i и j, и соедините новый узел с центральным узлом. Например, в части (B) рисунка справа узел u создается для соединения f и g.
- Рассчитайте расстояние от каждого таксона пары до этого нового узла.
- Рассчитайте расстояние от каждого таксона вне этой пары до нового узла.
- Запустите алгоритм еще раз, заменив пару соединенных соседей новым узлом и используя расстояния, рассчитанные на предыдущем шаге.



Q-матрица
[ редактировать ]На основе матрицы расстояний, связывающей таксоны, рассчитайте х матрица следующее:

| ( 1 ) |

где расстояние между таксонами и .



Расстояние от членов пары до нового узла
[ редактировать ]Для каждого таксона в объединяемой паре используйте следующую формулу для расчета расстояния до нового узла:
| ( 2 ) |
![{\displaystyle \delta (f,u)={\frac {1}{2}}d(f,g)+{\frac {1}{2(n-2)}}\left[\sum _{ k=1}^{n}d(f,k)-\sum _{k=1}^{n}d(g,k)\right]\quad }](https://wikimedia.org/api/rest_v1/media/math/render/svg/fbb5a4b2a6fbec48a4bbc6d6fc1e351e5eb851e0)
и:

Таксоны и являются парными таксонами и это вновь созданный узел. Ветви, соединяющиеся и и и , и их длины, и являются частью постепенно создаваемого дерева; они не влияют и не зависят от последующих шагов присоединения соседей.





Расстояние остальных таксонов от нового узла
[ редактировать ]Для каждого таксона, не учтенного на предыдущем шаге, мы рассчитываем расстояние до нового узла следующим образом:
| ( 3 ) |
![{\displaystyle d(u,k)={\frac {1}{2}}[d(f,k)+d(g,k)-d(f,g)]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/29d8e77a0543fb3258d08396a6fb44d387a92ad8)
где это новый узел, это узел, до которого мы хотим вычислить расстояние и и являются членами только что присоединившейся пары.

Сложность
[ редактировать ]Сосед присоединился к набору таксоны требуют итерации. На каждом этапе приходится строить и искать матрица. Первоначально матрица - это размер , то следующий шаг и т. д. Простая реализация этого приводит к алгоритму с временной сложностью ; [3] существуют реализации, которые используют эвристику, чтобы в среднем добиться гораздо большего результата. [4]




Пример
[ редактировать ]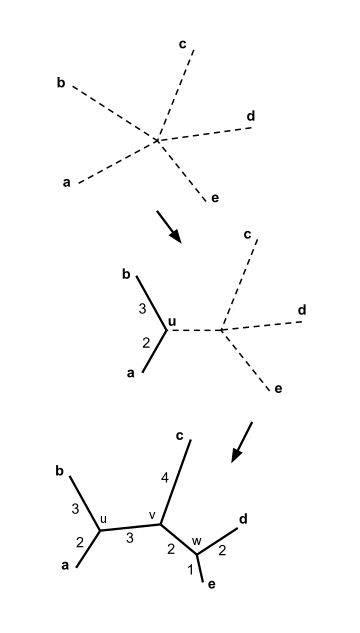
Предположим, что у нас есть пять таксонов. и следующая матрица расстояний :


| а | б | с | д | и | |
|---|---|---|---|---|---|
| а | 0 | 5 | 9 | 9 | 8 |
| б | 5 | 0 | 10 | 10 | 9 |
| с | 9 | 10 | 0 | 8 | 7 |
| д | 9 | 10 | 8 | 0 | 3 |
| и | 8 | 9 | 7 | 3 | 0 |
Первый шаг
[ редактировать ]Первое присоединение
[ редактировать ]Мы рассчитываем значения по уравнению ( 1 ). Например:



Мы получаем следующие значения для матрица (диагональные элементы матрицы не используются издесь опущено):
| а | б | с | д | и | |
|---|---|---|---|---|---|
| а | −50 | −38 | −34 | −34 | |
| б | −50 | −38 | −34 | −34 | |
| с | −38 | −38 | −40 | −40 | |
| д | −34 | −34 | −40 | −48 | |
| и | −34 | −34 | −40 | −48 |
В приведенном выше примере . Это наименьшее значение , поэтому мы соединяем элементы и .



Оценка длины первой ветки
[ редактировать ]Позволять обозначаем новый узел. Согласно уравнению ( 2 ), приведенному выше, ветви, соединяющие и к тогда имейте длину:
![{\displaystyle \delta (a,u)={\frac {1}{2}}d(a,b)+{\frac {1}{2(5-2)}}\left[\sum _{ k=1}^{5}d(a,k)-\sum _{k=1}^{5}d(b,k)\right]\quad ={\frac {5}{2}}+ {\frac {31-34}{6}}=2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/37ab7ad46b3277cb0d8fa097d54007520aadb1df)

Первое обновление матрицы расстояний
[ редактировать ]Затем мы приступаем к обновлению исходной матрицы расстояний. в новую матрицу расстояний (см. ниже), уменьшенный в размере на одну строку и один столбец за счет объединения с в своего соседа . Используя уравнение ( 3 ) выше, мы вычисляем расстояние от каждому из остальных узлов, кроме и . В этом случае мы получаем:

![{\displaystyle d(u,c)={\frac {1}{2}}[d(a,c)+d(b,c)-d(a,b)]={\frac {9+10 -5}{2}}=7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f4eedbd420cf866e0aca938b6b92ac75e7a332d)
![{\displaystyle d(u,d)={\frac {1}{2}}[d(a,d)+d(b,d)-d(a,b)]={\frac {9+10 -5}{2}}=7}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4c547916160e8c48cdf86f8c07bf9dc4fb079aa1)
![{\displaystyle d(u,e)={\frac {1}{2}}[d(a,e)+d(b,e)-d(a,b)]={\frac {8+9 -5}{2}}=6}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498737ead33eb4e18634803a7c2a940091865cb1)
Полученная матрица расстояний является:
| в | с | д | и | |
|---|---|---|---|---|
| в | 0 | 7 | 7 | 6 |
| с | 7 | 0 | 8 | 7 |
| д | 7 | 8 | 0 | 3 |
| и | 6 | 7 | 3 | 0 |
Жирные значения в соответствуют вновь рассчитанным расстояниям, тогда как значения, выделенные курсивом, не затрагиваются обновлением матрицы, поскольку они соответствуют расстояниям между элементами, не участвующими в первом объединении таксонов.
Второй шаг
[ редактировать ]Второе присоединение
[ редактировать ]Соответствующий матрица:

| в | с | д | и | |
|---|---|---|---|---|
| в | −28 | −24 | −24 | |
| с | −28 | −24 | −24 | |
| д | −24 | −24 | −28 | |
| и | −24 | −24 | −28 |
Мы можем выбрать либо присоединиться и или присоединиться и ; обе пары имеют минимальный ценность , и любой выбор приводит к одному и тому же результату. Для конкретики давайте присоединимся и и вызвать новый узел .





Оценка длины второй ветви
[ редактировать ]Длины соединяющихся ветвей и к можно рассчитать:
![{\displaystyle \delta (u,v)={\frac {1}{2}}d(u,c)+{\frac {1}{2(4-2)}}\left[\sum _{ k=1}^{4}d(u,k)-\sum _{k=1}^{4}d(c,k)\right]\quad ={\frac {7}{2}}+ {\frac {20-22}{4}}=3}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a469c064887abf42cd02ed9383f0fabff3d50379)

Соединение элементов и расчет длины ветвей помогают построить дерево соединения соседей, как показано на рисунке .
Обновление матрицы второго расстояния
[ редактировать ]Обновленная матрица расстояний для остальных 3 узлов, , , и , теперь вычисляется:

![{\displaystyle d(v,d)={\frac {1}{2}}[d(u,d)+d(c,d)-d(u,c)]={\frac {7+8 -7}{2}}=4}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9a149fca7dbf5339b8aaf177e55baf3ae19e3de6)
![{\displaystyle d(v,e)={\frac {1}{2}}[d(u,e)+d(c,e)-d(u,c)]={\frac {6+7 -7}{2}}=3}](https://wikimedia.org/api/rest_v1/media/math/render/svg/75942d802678c69bac9b44e8b213994e490fe44c)
| v | д | и | |
|---|---|---|---|
| v | 0 | 4 | 3 |
| д | 4 | 0 | 3 |
| и | 3 | 3 | 0 |
Последний шаг
[ редактировать ]На этом этапе древовидная топология полностью решена. Однако для наглядности можно вычислить матрица. Например:


| v | д | и | |
|---|---|---|---|
| v | −10 | −10 | |
| д | −10 | −10 | |
| и | −10 | −10 |
Для конкретики давайте присоединимся и и вызвать последний узел . Длины трех оставшихся ветвей можно вычислить:

![{\displaystyle \delta (v,w)={\frac {1}{2}}d(v,d)+{\frac {1}{2(3-2)}}\left[\sum _{ k=1}^{3}d(v,k)-\sum _{k=1}^{3}d(d,k)\right]\quad ={\frac {4}{2}}+ {\frac {7-7}{2}}=2}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ef24fb03c3726d7607adbd331c3301b26e40551)


Дерево объединения соседей теперь завершено, как показано на рисунке .
Вывод: аддитивные расстояния
[ редактировать ]Этот пример представляет собой идеализированный случай: обратите внимание, что если мы перейдем от одного таксона к любому другому по ветвям дерева и просуммируем длины пройденных ветвей, результат равен расстоянию между этими таксонами во входной матрице расстояний. Например, переходя от к у нас есть . Матрица расстояний, расстояния которой таким образом согласуются с некоторым деревом, называется «аддитивной», что на практике встречается редко. Тем не менее важно отметить, что, если в качестве входных данных используется аддитивная матрица расстояний, объединение соседей гарантированно найдет дерево, расстояния между таксонами которого совпадают с ним.

Присоединение соседей как минимальная эволюция
[ редактировать ]Присоединение соседей можно рассматривать как жадную эвристику для сбалансированной минимальной эволюции. [5] (BME) критерий. Для каждой топологии BME определяет длину дерева (сумму длин ветвей) как определенную взвешенную сумму расстояний в матрице расстояний, причем веса зависят от топологии. Оптимальной топологией BME является та, которая минимизирует длину этого дерева. Нью-Джерси на каждом этапе жадно объединяет ту пару таксонов, которая даст наибольшее уменьшение расчетной длины дерева. Эта процедура не гарантирует нахождение оптимума для критерия BME, хотя часто дает такой результат и обычно оказывается довольно близким к нему. [5]
Преимущества и недостатки
[ редактировать ]Главное достоинство Нью-Джерси в том, что он быстрый. [6] : 466 по сравнению с наименьших квадратов , максимальной экономии и максимального правдоподобия . методами [6] Это делает его практичным для анализа больших наборов данных (сотни или тысячи таксонов) и для начальной загрузки , для которых другие средства анализа (например, максимальная экономия , максимальное правдоподобие ) могут оказаться непомерно вычислительными .
Соединение соседей обладает тем свойством, что если входная матрица расстояний правильна, то и выходное дерево будет правильным.Более того, корректность топологии выходного дерева гарантируется до тех пор, пока матрица расстояний является «почти аддитивной», особенно если каждая запись в матрице расстояний отличается от истинного расстояния менее чем на половину длины кратчайшей ветви дерева. [7] На практике матрица расстояний редко удовлетворяет этому условию, но объединение соседей все равно часто создает правильную топологию дерева. [8] Корректность объединения соседей для почти аддитивных матриц расстояний подразумевает, что оно статистически согласовано во многих моделях эволюции; при наличии данных достаточной длины объединение соседей с высокой вероятностью восстановит истинное дерево.По сравнению с UPGMA и WPGMA , объединение соседей имеет то преимущество, что оно не предполагает, что все линии развиваются с одинаковой скоростью ( гипотеза молекулярных часов ).
Тем не менее, соединение соседей в значительной степени вытеснено филогенетическими методами, которые не полагаются на измерения расстояний и обеспечивают превосходную точность в большинстве условий. [ нужна ссылка ] Объединение соседей имеет нежелательную особенность: оно часто присваивает некоторым ветвям отрицательную длину.
Реализации и варианты
[ редактировать ]Существует множество программ, реализующих присоединение к соседям. Среди реализаций канонического NJ (т. е. использующего классические критерии оптимизации NJ и, следовательно, дающего те же результаты), RapidNJ (начат в 2003 г., основное обновление в 2011 г., все еще обновляется в 2023 г.) [9] и NINJA (начало 2009 г., последнее обновление 2013 г.) [10] считаются современными. Типичное время их выполнения пропорционально квадрату числа таксонов.
Варианты, отклоняющиеся от канонических, включают:
- БИОНДЖ (1997) [11] и Вейбор (2000), [12] повышение точности за счет использования того факта, что более короткие расстояния в матрице расстояний обычно известны лучше, чем более длинные. Оба метода были расширены для работы с неполными матрицами расстояний. [13]
- «Быстрый Нью-Джерси» запоминает лучший узел и всегда равен O(n^2); «Relax NJ» выполняет поиск с подъемом на холм и сохраняет сложность наихудшего случая O(n^3). Рапид Нью-Джерси быстрее, чем обычный расслабленный Нью-Джерси. [14]
- FastME — это реализация тесно связанного метода сбалансированной минимальной эволюции (BME) (см. § Соединение соседей как минимальная эволюция ). Он примерно такой же быстрый и более точный, чем Нью-Джерси. Он начинается с грубого дерева, а затем улучшает его с помощью набора топологических ходов, таких как обмен ближайшими соседями (NNI). [15] FastTree — родственный метод. Он работает с «профилями» последовательности, а не с матрицей. Он начинается с дерева примерно Нью-Джерси, преобразует его в BME, а затем преобразует в приближенное максимальное правдоподобие. [16]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Сайто, Н.; Ней, М. (1 июля 1987 г.). «Метод объединения соседей: новый метод реконструкции филогенетических деревьев» . Молекулярная биология и эволюция . 4 (4): 406–425. doi : 10.1093/oxfordjournals.molbev.a040454 . ПМИД 3447015 .
- ^ Ксавье Дидло (2010). «Последовательный анализ структур бактериальных популяций» . В Д. Эшли Робинсон; Дэниел Фалуш; Эдвард Дж. Фейл (ред.). Бактериальная популяционная генетика при инфекционных заболеваниях . Джон Уайли и сыновья. стр. 46–47. ISBN 978-0-470-42474-2 .
- ^ Стьюдер, Дж. А.; Кепплер, К.Дж. (ноябрь 1988 г.). «Заметка об алгоритме соединения соседей Сайто и Ней» . Молекулярная биология и эволюция . 5 (6): 729–31. doi : 10.1093/oxfordjournals.molbev.a040527 . ISSN 1537-1719 . ПМИД 3221794 .
- ^ Майлунд, Томас; Бродал, ГертС; Фагерберг, Рольф; Педерсен, КристианНС; Филлипс, Дерек (2006). «Переработка метода соединения соседей» . БМК Биоинформатика . 7 (1): 29. дои : 10.1186/1471-2105-7-29 . ПМЦ 3271233 . ПМИД 16423304 .
- ^ Jump up to: Перейти обратно: а б Гаскуэль О, Стил М (2006). «Обнаружено присоединение соседей» . Мол Биол Эвол . 23 (11): 1997–2000. дои : 10.1093/molbev/msl072 . ПМИД 16877499 .
- ^ Jump up to: Перейти обратно: а б Кунер, МК; Фельзенштейн, Дж. (1 мая 1994 г.). «Сравнение моделирования алгоритмов филогении при равных и неравных скоростях эволюции» . Молекулярная биология и эволюция . 11 (3): 459–468. doi : 10.1093/oxfordjournals.molbev.a040126 . ISSN 0737-4038 . ПМИД 8015439 .
- ^ Аттесон К. (1997). «Производительность алгоритмов объединения соседей реконструкции филогении», стр. 101–110. Цзян Т. и Ли Д., ред., Конспекты лекций по информатике, 1276 , Springer-Verlag, Берлин. КОКООН '97.
- ^ Михаеску Р., Леви Д., Пахтер Л. (2009). «Почему объединение соседей работает». Алгоритмика . 54 (1): 1–24. arXiv : cs/0602041 . дои : 10.1007/s00453-007-9116-4 . S2CID 2462145 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ «РапидНЖ» . birc.au.dk.
- ^ «NINJA: инструмент для крупномасштабного филогенетического вывода по объединению соседей - Домой» . Wheelerlab.org .
- ^ «АТГК: БиоНЖ» . www.atgc-montpellier.fr .
- ^ «Домашняя страница WEIGHBOR» . 5 марта 2015 г. Архивировано из оригинала 05 марта 2015 г.
- ^ Крискуоло, Алексис; Гаскюэль, Оливье (декабрь 2008 г.). «Быстрые алгоритмы типа Нью-Джерси для работы с неполными матрицами расстояний» . БМК Биоинформатика . 9 (1): 166. дои : 10.1186/1471-2105-9-166 . ПМК 2335114 . ПМИД 18366787 .
- ^ Симонсен, Мартин; Майлунд, Томас; Педерсен, Кристиан Н.С. (2008). «Быстрое присоединение к соседям» (PDF) . Алгоритмы в биоинформатике . Конспекты лекций по информатике. Том. 5251. стр. 113–122. дои : 10.1007/978-3-540-87361-7_10 . ISBN 978-3-540-87360-0 .
- ^ «АТГК: FastME» . www.atgc-montpellier.fr .
- ^ «FastTree 2.1: Деревья приближения максимального правдоподобия для больших трасс» . www.microbesonline.org .
Другие источники
[ редактировать ]- Студент Дж. А., Кепплер К. Дж. (1988). «Заметка об алгоритме соединения соседей Сайто и Ней» . Мол Биол Эвол . 5 (6): 729–731. doi : 10.1093/oxfordjournals.molbev.a040527 . ПМИД 3221794 .
- Мартин Симонсен; Томас Майлунд; Кристиан Н.С. Педерсен (2008). «Быстрое присоединение к соседям». Алгоритмы в биоинформатике . Конспекты лекций по информатике. Том. 5251. стр. 113–122. CiteSeerX 10.1.1.218.2078 . дои : 10.1007/978-3-540-87361-7_10 . ISBN 978-3-540-87360-0 .
Внешние ссылки
[ редактировать ]- Метод присоединения соседей — учебное пособие
| Соответствующие поля | ||
|---|---|---|
| Основные понятия | ||
| Методы вывода | ||
| Текущие темы | ||
| Групповые черты | ||
| Типы групп | ||
| Номенклатура | ||