Секвенирование экзома

Секвенирование экзома , также известное как секвенирование всего экзома ( WES ), представляет собой геномный метод секвенирования всех белково-кодирующих областей генов в геноме (известном как экзом ). [ 1 ] Он состоит из двух этапов: первый шаг — выбрать только ту часть ДНК , которая кодирует белки . Эти области известны как экзоны — у людей около 180 000 экзонов, составляющих около 1% человеческого генома , или примерно 30 миллионов пар оснований . Вторым шагом является секвенирование экзонной ДНК с использованием любой технологии высокопроизводительного секвенирования ДНК . [ 2 ]
Целью этого подхода является выявление генетических вариантов, которые изменяют последовательности белков, и сделать это с гораздо меньшими затратами, чем полногеномное секвенирование . Поскольку эти варианты могут быть ответственны как за менделевские , так и за распространенные полигенные заболевания, такие как болезнь Альцгеймера , полное секвенирование экзома применялось как в академических исследованиях, так и в качестве клинической диагностики.

Мотивация и сравнение с другими подходами
[ редактировать ]Секвенирование экзома особенно эффективно при изучении редких менделевских заболеваний, поскольку это эффективный способ идентифицировать генетические варианты во всех генах человека. Эти заболевания чаще всего вызываются очень редкими генетическими вариантами, которые присутствуют лишь у небольшого числа людей; [ 3 ] напротив, такие методы, как массивы SNP, могут обнаруживать только общие генетические варианты, которые являются общими для многих людей в более широкой популяции. [ 4 ] Более того, поскольку варианты, вызывающие тяжелые заболевания, с гораздо большей вероятностью (но ни в коем случае не исключительно) находятся в кодирующей последовательности белка, [ 5 ] [ 6 ] сосредоточение внимания на этом 1% обходится гораздо дешевле, чем секвенирование всего генома, но при этом позволяет обнаружить высокий выход соответствующих вариантов.
В прошлом клинические генетические тесты выбирались на основе клинической картины пациента (т.е. фокусировались на одном гене или небольшом количестве генов, которые, как известно, связаны с определенным синдромом), или исследовали только определенные типы вариаций (например, сравнительная геномная гибридизация ). но обеспечил окончательный генетический диагноз менее чем у половины всех пациентов. [ 7 ] Секвенирование экзома в настоящее время все чаще используется в дополнение к другим тестам: как для поиска мутаций в генах, которые, как известно, вызывают заболевание, так и для выявления новых генов путем сравнения экзомов у пациентов со схожими характеристиками. [ нужна ссылка ]
Техническая методология
[ редактировать ]Шаг 1: Стратегии целевого обогащения
[ редактировать ]Методы целевого обогащения позволяют выборочно захватывать интересующие геномные области из образца ДНК перед секвенированием. С момента первоначального описания метода прямой геномной селекции (DGS) в 2005 году было разработано несколько стратегий целевого обогащения. [ 8 ]
Хотя для целевого захвата было описано множество методов, лишь некоторые из них были расширены для захвата целых экзомов. [ 9 ] Первой стратегией целевого обогащения, которая была применена к секвенированию всего экзома, был метод гибридного захвата на основе массива в 2007 году, но в последние годы приобрел популярность захват в растворе.
Захват на основе массива
[ редактировать ]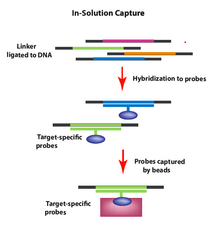
Микрочипы содержат одноцепочечные олигонуклеотиды с последовательностями из генома человека, образующие мозаику интересующей области, фиксированную на поверхности. Геномная ДНК разрезается с образованием двухцепочечных фрагментов. Фрагменты подвергаются ремонту концов, образуя тупые концы, и добавляются адаптеры с универсальными последовательностями прайминга. Эти фрагменты гибридизуются с олигонуклеотидами на микрочипе. Негибридизованные фрагменты смываются, а желаемые фрагменты элюируются. Затем фрагменты амплифицируют с помощью ПЦР . [ 10 ] [ 11 ]
Компания Roche NimbleGen первой применила оригинальную технологию DGS. [ 8 ] и адаптировать его для секвенирования следующего поколения. Они разработали массив Sequence Capture Human Exome 2.1M Array, позволяющий захватывать около 180 000 кодирующих экзонов. [ 12 ] Этот метод экономит время и экономически эффективен по сравнению с методами, основанными на ПЦР. Матрица захвата Agilent и массив сравнительной геномной гибридизации — это другие методы, которые можно использовать для гибридного захвата целевых последовательностей. Ограничения этого метода включают необходимость дорогостоящего оборудования, а также относительно большого количества ДНК. [ 13 ]
Захват в растворе
[ редактировать ]пул индивидуальных олигонуклеотидов Для захвата интересующих геномных областей с помощью захвата в растворе синтезируется (зондов) и гибридизуется в растворе с образцом фрагментированной геномной ДНК. Зонды (помеченные шариками) избирательно гибридизуются с интересующими областями генома, после чего шарики (теперь содержащие интересующие фрагменты ДНК) можно извлечь и промыть, чтобы удалить излишки материала. Затем шарики удаляют, и геномные фрагменты можно секвенировать, что позволяет избирательно секвенировать ДНК интересующих геномных областей (например, экзонов).
Этот метод был разработан для улучшения метода целевого обогащения гибридизационного захвата. При захвате раствора (в отличие от гибридного захвата) количество зондов для целевых областей, представляющих интерес, превышает необходимое количество шаблона. [ 13 ] Оптимальный целевой размер составляет около 3,5 мегабаз и обеспечивает превосходный охват последовательностей целевых регионов. Предпочтительный метод зависит от нескольких факторов, в том числе: количества пар оснований в интересующей области, требований к целевому считыванию, наличия собственного оборудования и т. д. [ 14 ]
Шаг 2: Секвенирование
[ редактировать ]Существует множество платформ секвенирования следующего поколения , устаревших классических методологий секвенирования Сэнгера. Другие платформы включают секвенатор Roche 454 и Life Technologies системы SOLiD, Life Technologies Ion Torrent и Illumina Genome Analyser II (несуществующий), а также последующие инструменты серий Illumina MiSeq, HiSeq и NovaSeq, все из которых можно использовать для массово-параллельного секвенирования экзома. Эти системы NGS с «коротким считыванием» особенно хорошо подходят для анализа многих относительно коротких участков последовательности ДНК, обнаруженных в экзонах человека.
Сравнение с другими технологиями
[ редактировать ]Существует множество технологий, позволяющих идентифицировать генетические варианты. Каждая технология имеет преимущества и недостатки с точки зрения технических и финансовых факторов. Двумя такими технологиями являются микрочипы и полногеномное секвенирование .
Генотипирование на основе микрочипов
[ редактировать ]Микрочипы используют зонды гибридизации для проверки распространенности известных последовательностей ДНК, поэтому их нельзя использовать для выявления неожиданных генетических изменений. [ 13 ] Напротив, технологии высокопроизводительного секвенирования, используемые при секвенировании экзома, напрямую обеспечивают нуклеотидные последовательности ДНК в тысячах протестированных экзонных локусов. [ 15 ] Таким образом, WES устраняет некоторые из существующих ограничений массивов гибридного генотипирования .
Хотя секвенирование экзома обходится дороже, чем технологии, основанные на гибридизации, в расчете на образец, его стоимость снижается из-за снижения стоимости и увеличения производительности секвенирования всего генома . [ нужна ссылка ]
Полногеномное секвенирование
[ редактировать ]Секвенирование экзома позволяет идентифицировать только те варианты, обнаруженные в кодирующей области генов, которые влияют на функцию белка. Он не способен идентифицировать структурные и некодирующие варианты, связанные с заболеванием, которые можно найти с помощью других методов, таких как полногеномное секвенирование . [ 2 ] Остается 99% генома человека, не покрытого секвенированием экзома, а секвенирование экзома позволяет секвенировать части генома как минимум в 20 раз больше образцов по сравнению с секвенированием всего генома. [ 2 ] Что касается перевода выявленных редких вариантов в клинику, размер выборки и способность интерпретировать результаты для постановки клинического диагноза указывают на то, что при нынешних знаниях в области генетики имеются сообщения об использовании секвенирования экзома для помощи в диагностике. [ 12 ] Стоимость секвенирования экзома обычно ниже, чем стоимость секвенирования всего генома. [ 16 ]
Анализ данных
[ редактировать ]Статистический анализ большого количества данных, полученных с помощью методов секвенирования, является сложной задачей. Даже при секвенировании экзомов отдельных людей генерируется большое количество данных и информации о последовательностях, что требует значительного объема анализа данных. Проблемы, связанные с анализом этих данных, включают изменения в программах, используемых для выравнивания и сборки считываний последовательностей. [ 13 ] Различные технологии секвенирования также имеют разную частоту ошибок и генерируют различную длину считывания, что может создавать проблемы при сравнении результатов, полученных на разных платформах секвенирования.
Ложноположительные и ложноотрицательные результаты связаны с подходами к повторному секвенированию генома и являются критическими проблемами. Для улучшения качества данных экзома было разработано несколько стратегий, таких как:
- Сравнение генетических вариантов, выявленных при секвенировании и генотипировании на основе массивов [ 2 ]
- Сравнение кодирующих SNP с секвенированным полным геномом человека с заболеванием [ 2 ]
- Сравнение кодирующих SNP с секвенированием по Сэнгеру особей HapMap [ 2 ]
Редкие рецессивные заболевания могут не иметь однонуклеотидных полиморфизмов (SNP) в общедоступных базах данных, таких как dbSNP . Более распространенные рецессивные фенотипы с большей вероятностью будут иметь варианты, вызывающие заболевания, о которых сообщается в dbSNP. Например, наиболее распространенный вариант муковисцидоза имеет частоту аллелей около 3% в большинстве популяций. Отсев таких вариантов может ошибочно исключить такие гены из рассмотрения. Гены рецессивных заболеваний обычно легче идентифицировать, чем доминантные расстройства, поскольку гены с меньшей вероятностью будут иметь более одного редкого несинонимичного варианта. [ 2 ] Система, проверяющая распространенные генетические варианты, опирается на dbSNP, который может не иметь точной информации о вариациях аллелей. Использование списков общих вариаций из исследуемого экзома или полногеномного секвенированного человека было бы более надежным. Проблема этого подхода заключается в том, что по мере увеличения количества секвенированных экзомов количество необычных вариантов dbSNP также будет увеличиваться. Будет необходимо разработать пороговые значения для определения распространенных вариантов, которые вряд ли будут связаны с фенотипом заболевания. [ 15 ]
Генетическая гетерогенность и этническая принадлежность населения также являются серьезными ограничениями, поскольку они могут увеличить количество ложноположительных и ложноотрицательных результатов, что затруднит идентификацию генов-кандидатов. Конечно, можно снизить жесткость пороговых значений при наличии гетерогенности и этнической принадлежности, однако это также снизит возможность обнаружения вариантов. Использование подхода «сначала генотип» для идентификации генов-кандидатов также может предложить решение для преодоления этих ограничений.
В отличие от анализа распространенных вариантов, анализ редких вариантов в исследованиях секвенирования всего экзома оценивает наборы вариантов, а не отдельные варианты. [ 17 ] [ 18 ] Функциональные аннотации прогнозируют эффект или функцию редких вариантов и помогают расставить приоритеты редких функциональных вариантов. Включение этих аннотаций может эффективно повысить эффективность генетической ассоциации анализа редких вариантов в исследованиях полногеномного секвенирования. [ 19 ] Были разработаны некоторые методы и инструменты для проведения функционально обоснованного анализа ассоциаций редких вариантов путем включения функциональных аннотаций, расширяющих возможности анализа в исследованиях секвенирования всего экзома. [ 20 ] [ 21 ]
Этические последствия
[ редактировать ]Новые технологии в геномике изменили подход исследователей как к фундаментальным, так и к трансляционным исследованиям. С помощью таких подходов, как секвенирование экзома, можно значительно улучшить данные, полученные из отдельных геномов, что поставило ряд вопросов о том, как обращаться с огромным объемом информации. Следует ли предоставить участникам этих исследований доступ к информации о секвенировании? Следует ли передавать эту информацию страховым компаниям? Эти данные могут привести к неожиданным результатам и усложнить клиническую полезность и пользу для пациентов. Эта область геномики по-прежнему остается сложной задачей, и исследователи ищут способы решения этих вопросов. [ 15 ]
Применение секвенирования экзома
[ редактировать ]Используя секвенирование экзома, исследования с фиксированной стоимостью могут секвенировать образцы на гораздо большую глубину, чем это можно было бы достичь при полногеномном секвенировании. Эта дополнительная глубина делает секвенирование экзома хорошо подходящим для ряда приложений, требующих надежных вызовов вариантов.
Картирование редких вариантов при сложных заболеваниях
[ редактировать ]Текущие исследования ассоциаций сосредоточены на общих вариациях по всему геному, поскольку их легче всего выявить с помощью наших текущих анализов. Однако в исследованиях генов-кандидатов было обнаружено, что вызывающие заболевания варианты с большим эффектом лежат в экзомах и из-за отрицательного отбора обнаруживаются с гораздо более низкими частотами аллелей и могут оставаться нетипизированными в текущих стандартных анализах генотипирования. Полногеномное секвенирование является потенциальным методом анализа новых вариантов генома. Однако считается, что при сложных расстройствах (таких как аутизм) большое количество генов связано с риском заболевания. [ 1 ] [ 22 ] Эта гетерогенность основного риска означает, что для открытия генов требуются очень большие размеры выборки, и, следовательно, полногеномное секвенирование не особенно экономически эффективно. Проблема размера выборки решается благодаря разработке новых передовых аналитических методов, которые эффективно картируют гены болезней, несмотря на то, что генетические мутации на уровне вариантов редки. [ 22 ] Кроме того, варианты в кодирующих регионах были гораздо более тщательно изучены, и их функциональное значение гораздо легче выявить, что делает практическое применение вариантов в целевой области экзома более доступным.
Секвенирование экзома при открытии редких вариантов генов остается очень активной и продолжающейся областью исследований, и появляется все больше свидетельств того, что значительное бремя риска наблюдается для наборов генов. Сообщалось о редких вариантах гена KRT82 при секвенировании экзома при аутоиммунном заболевании очаговой алопеции. [ 1 ]
Открытие менделевских расстройств
[ редактировать ]Результаты, полученные к настоящему времени, свидетельствуют о том, что при менделевских расстройствах с большим эффектом в основе всего заболевания лежит один или очень небольшое количество вариантов кодирующих генов. Из-за серьезности этих нарушений предполагается, что несколько причинных вариантов являются чрезвычайно редкими или новыми для популяции и могут быть пропущены любым стандартным анализом генотипирования. Секвенирование экзома обеспечивает вызов вариантов с высоким охватом в регионах кодирования, которые необходимы для отделения истинных вариантов от шума. Успешная модель открытия менделевских генов предполагает открытие вариантов de novo с использованием трио-секвенирования, при котором генотипируются родители и пробанд.
Тематические исследования
[ редактировать ]В исследовании, опубликованном в сентябре 2009 года, обсуждался эксперимент, подтверждающий концептуальный эксперимент, чтобы определить, можно ли идентифицировать причинные генетические варианты с помощью секвенирования экзома. Они секвенировали четырех человек с синдромом Фримена-Шелдона (FSS) (OMIM 193700), редким аутосомно-доминантным заболеванием, которое, как известно, вызвано мутацией в гене MYH3 . [ 2 ] Восемь особей HapMap также были секвенированы для удаления общих вариантов и идентификации причинного гена FSS. После исключения распространенных вариантов авторы смогли идентифицировать MYH3 , что подтверждает, что секвенирование экзома можно использовать для выявления причинных вариантов редких заболеваний. [ 2 ] Это было первое опубликованное исследование, в котором секвенирование экзома использовалось в качестве подхода к идентификации неизвестного гена, вызывающего редкое менделевское расстройство.
Впоследствии другая группа сообщила об успешной клинической диагностике пациента с подозрением на синдром Бартера турецкого происхождения. [ 12 ] Синдром Барттера – заболевание, связанное с потерей солей в почках. Секвенирование экзома выявило неожиданную хорошо консервативную рецессивную мутацию в гене под названием SLC26A3, который связан с врожденной хлоридной диареей (CLD). Молекулярный диагноз ХЗЛ был подтвержден лечащим врачом. Этот пример предоставил доказательство концепции использования секвенирования всего экзома в качестве клинического инструмента при оценке пациентов с невыявленными генетическими заболеваниями. Этот отчет считается первым применением технологии секвенирования нового поколения для молекулярной диагностики пациента.
Второй отчет был посвящен секвенированию экзома у людей с менделевским расстройством, известным как синдром Миллера (MIM#263750), редким заболеванием аутосомно-рецессивного наследования. Были изучены два брата и сестры и два неродственных человека с синдромом Миллера. Они рассмотрели варианты, которые потенциально могут быть патогенными, такие как несинонимичные мутации, акцепторные и донорские сайты сплайсинга, а также вставки или делеции короткого кодирования. [ 3 ] Поскольку синдром Миллера является редким заболеванием, ожидается, что его причинный вариант ранее не был идентифицирован. Предыдущие исследования секвенирования экзома распространенных однонуклеотидных полиморфизмов (SNP) в общедоступных базах данных SNP были использованы для дальнейшего исключения генов-кандидатов. После исключения этих генов авторы обнаружили мутации DHODH , которые были распространены среди людей с синдромом Миллера. Каждый человек с синдромом Миллера был сложной гетерозиготой по мутациям DHODH , которые передавались по наследству, поскольку каждый родитель больного человека оказался носителем. [ 3 ]
Впервые было показано, что секвенирование экзома идентифицирует новый ген, ответственный за редкое менделевское заболевание. Это захватывающее открытие демонстрирует, что секвенирование экзома потенциально способно обнаружить гены, вызывающие сложные заболевания, что ранее было невозможно из-за ограничений традиционных методов. Целенаправленный захват и массовое параллельное секвенирование представляют собой экономически эффективную, воспроизводимую и надежную стратегию с высокой чувствительностью и специфичностью для обнаружения вариантов, вызывающих изменения кодирования белков в отдельных геномах человека.
Клиническая диагностика
[ редактировать ]Секвенирование экзома можно использовать для диагностики генетической причины заболевания у пациента. Выявление мутаций гена, лежащего в основе заболевания, может иметь важное значение для диагностических и терапевтических подходов, может помочь в прогнозировании естественного течения заболевания и позволяет тестировать членов семьи из группы риска. [ 2 ] [ 3 ] [ 12 ] [ 23 ] [ 24 ] [ 25 ] Существует множество факторов, которые делают секвенирование экзома превосходящим анализ одного гена, включая способность выявлять мутации в генах, которые не были проверены из-за атипичной клинической картины. [ 25 ] или способность выявлять клинические случаи, когда мутации разных генов способствуют разным фенотипам у одного и того же пациента. [ 3 ]
После диагностики генетической причины заболевания эта информация может помочь в выборе соответствующего лечения. Впервые эта стратегия была успешно применена в клинике при лечении ребенка с воспалительным заболеванием кишечника. [ 24 ] [ 26 ] Ранее использовался ряд традиционных методов диагностики, но результаты не могли объяснить симптомы ребенка. Анализ данных секвенирования экзома выявил мутацию в гене XIAP . Знание функции этого гена определило лечение младенца, что привело к трансплантации костного мозга, которая излечила ребенка от болезни. [ 24 ]
Исследователи использовали секвенирование экзома, чтобы определить основную мутацию у пациента с синдромом Бартера и врожденной хлоридной диареей. [ 12 ] Группа Билгуляра также использовала секвенирование экзома и определила основную мутацию у пациента с тяжелыми пороками развития головного мозга, заявив: «[Эти результаты] подчеркивают использование полного секвенирования экзома для выявления локусов заболевания в условиях, в которых традиционные методы оказались сложными... Наши результаты продемонстрировать, что эта технология будет особенно ценна для открытия генов в тех условиях, в которых картирование затрудняется гетерогенностью локусов и неопределенностью границ диагностической классификации, что указывает на светлое будущее ее широкого применения в медицине». [ 23 ]
Исследователи из Университета Кейптауна, Южная Африка, использовали секвенирование экзома, чтобы обнаружить генетическую мутацию CDH2 как основную причину генетического нарушения, известного как аритмогенная кардиомиопатия правого желудочка (АРКП), которая увеличивает риск сердечных заболеваний и остановки сердца. [1]
Коммерческие затраты
[ редактировать ]Многие компании предложили потребителям секвенирование экзома. Knome была первой компанией, предложившей потребителям услуги по секвенированию экзома. [ когда? ] , стоимостью в несколько тысяч долларов. [ 27 ] Позже 23andMe запустила пилотную программу WES, о которой было объявлено в сентябре 2011 года и которая была прекращена в 2012 году. Потребители могли получить данные exome по цене 999 долларов. Компания предоставила необработанные данные и не предложила анализа. [ 27 ] [ 28 ] [ 29 ]
В ноябре 2012 года DNADTC, подразделение Gene by Gene, начало предлагать экзомы с 80-кратным покрытием и начальной ценой 695 долларов. [ 30 ] Эта цена за веб-сайт DNADTC в настоящее время составляет 895 долларов США. В октябре 2013 года BGI объявила о рекламной акции на личное секвенирование всего экзома с 50-кратным охватом за 499 долларов. [ 31 ] В июне 2016 года компания Genos смогла добиться еще более низкой цены в 399 долларов за сертифицированный CLIA потребительский экзом 75X, секвенированный из слюны. [ 32 ] [ 33 ] [ 34 ]
Обзор 36 исследований, проведенный в 2018 году, показал, что стоимость секвенирования экзома варьируется от 555 до 5169 долларов США, а диагностическая эффективность варьируется от 3% до 79% в зависимости от групп пациентов. [ 16 ]
См. также
[ редактировать ]- Профилирование ДНК
- Генетическое консультирование
- Персонализированная медицина
- Транскриптомика
- Полногеномное секвенирование
Ссылки
[ редактировать ]- ^ Jump up to: а б с Эрьявец С.О., Гельфман С., Абдельазиз А.Р., Ли Э.Ю., Монга И., Алкелай А., Ионита-Лаза И., Петухова Л., Кристиано А.М. (февраль 2022 г.). «Полное секвенирование экзома при очаговой алопеции выявляет редкие варианты KRT82» . Нат Коммун . 13 (1): 800. Бибкод : 2022NatCo..13..800E . дои : 10.1038/s41467-022-28343-3 . ПМЦ 8831607 . ПМИД 35145093 .
- ^ Jump up to: а б с д и ж г час я дж Нг С.Б., Тернер Э.Х., Робертсон П.Д., Флайгэр С.Д., Бигэм А.В., Ли С., Шаффер Т., Вонг М., Бхаттачарджи А., Эйхлер Э.Э., Бамшад М., Никерсон Д.А., Шендуре Дж. (10 сентября 2009 г.). «Целевой захват и массовое параллельное секвенирование 12 экзомов человека» . Природа . 461 (7261): 272–276. Бибкод : 2009Natur.461..272N . дои : 10.1038/nature08250 . ПМЦ 2844771 . ПМИД 19684571 .
- ^ Jump up to: а б с д и Сара Б. Нг; Кэти Дж. Бэкингем; Чоли Ли; Эбигейл В. Бигхэм; Холли К. Табор; Карин М Дент; Чад Д. Хафф; Пол Т. Шеннон; Этилин Ван Джабс; Дебора А. Никерсон; Джей Шендюр; Майкл Дж. Бэмшад (2010). «Секвенирование экзома выявляет причину менделевского расстройства» . Природная генетика . 42 (1): 30–35. дои : 10.1038/ng.499 . ПМЦ 2847889 . ПМИД 19915526 .
- ^ Ван, генеральный директор; Фан, Дж.Б.; Сяо, CJ; Берно, А.; Янг, П.; Сапольский, Р.; Гандур, Г.; Перкинс, Н.; Винчестер, Э. (15 мая 1998 г.). «Крупномасштабная идентификация, картирование и генотипирование однонуклеотидных полиморфизмов в геноме человека». Наука . 280 (5366): 1077–1082. Бибкод : 1998Sci...280.1077W . дои : 10.1126/science.280.5366.1077 . ISSN 0036-8075 . ПМИД 9582121 .
- ^ Петерсен, Бритт-Сабина; Фредрих, Бродер; Хоппнер, Марк П.; Эллингхаус, Дэвид; Франке, Андре (14 февраля 2017 г.). «Возможности и проблемы полногеномного и -экзомного секвенирования» . БМК Генетика . 18 (1): 14. дои : 10.1186/s12863-017-0479-5 . ISSN 1471-2156 . ПМК 5307692 . ПМИД 28193154 .
- ^ Ботштейн, Дэвид; Риш, Нил (март 2003 г.). «Обнаружение генотипов, лежащих в основе фенотипов человека: прошлые успехи в лечении менделевской болезни, будущие подходы к сложным заболеваниям» . Природная генетика . 33 (3): 228–237. дои : 10.1038/ng1090 . ISSN 1546-1718 . ПМИД 12610532 . S2CID 10599219 .
- ^ Раух, А; Хойер, Дж; Гут, С; Цвайер, К; Краус, К; Беккер, К; Ценкер, М; Хюффмайер, Ю; Тиль, К; Рюшендорф, Ф; Нюрнберг, П; Рейс, А; Траутманн, Ю (1 октября 2006 г.). «Диагностическая эффективность различных генетических подходов у пациентов с необъяснимой задержкой развития или умственной отсталостью» . Американский журнал медицинской генетики, часть A. 140 (19): 2063–74. дои : 10.1002/ajmg.a.31416 . ПМИД 16917849 . S2CID 24570999 .
- ^ Jump up to: а б Ставрос Басиардес; Роуз Вейл; Синди Хелмс; Элейн Р. Мардис; Энн М. Боукок; Майкл Ловетт (2005). «Прямой геномный отбор». Природные методы . 1 (2): 63–69. дои : 10.1038/nmeth0105-63 . ПМИД 16152676 . S2CID 667227 .
- ^ Тир, Дж. К.; Малликин, Дж. К. (12 августа 2010 г.). «Секвенирование экзома: золотая середина перед целыми геномами» . Молекулярная генетика человека . 19 (С2): Р145–Р151. дои : 10.1093/hmg/ddq333 . ПМЦ 2953745 . ПМИД 20705737 .
- ^ Эмили Х. Тернер; Сара Б. Нг; Дебора А. Никерсон; Джей Шендур (2009). «Методы разделения генома». Анну Рев Геном Хум Генет . 10 (1): 30–35. doi : 10.1146/annurev-genom-082908-150112 . ПМИД 19630561 .
- ^ Мертес Ф., Эльшарави А., Зауэр С., ван Хелворт Дж.М., ван дер Зааг П.Дж., Франке А., Нильссон М., Лерах Х., Брукс А.Дж. (2011). «Целевое обогащение участков геномной ДНК для секвенирования нового поколения» . Краткий. Функц. Геномика . 10 (6): 374–386. дои : 10.1093/bfgp/elr033 . ПМЦ 3245553 . ПМИД 22121152 . Архивировано из оригинала (PDF) 21 сентября 2017 г. Проверено 24 октября 2018 г.
- ^ Jump up to: а б с д и Чой М., Шолл У.И., Джи В., Лю Т., Тихонова И.Р., Зумбо П., Наир А., Баккалоглу А., Озен С., Санджад С., Нельсон-Уильямс С., Фархи А., Мане С., Лифтон Р.П. (10 ноября 2009 г.). «Генетическая диагностика путем захвата всего экзома и массового параллельного секвенирования ДНК» . Proc Natl Acad Sci США . 106 (45): 19096–19101. Бибкод : 2009PNAS..10619096C . дои : 10.1073/pnas.0910672106 . ПМЦ 2768590 . ПМИД 19861545 .
- ^ Jump up to: а б с д Кахведжян А., Квакенбуш Дж., Томпсон Дж. Ф. (2008). «Что бы вы сделали, если бы могли все упорядочить?» . Природная биотехнология . 26 (10): 1125–1133. дои : 10.1038/nbt1494 . ПМЦ 4153598 . ПМИД 18846086 .
- ^ Лира Маманова; Коффи, Элисон Дж; Скотт, Кэрол Э; Козарева, Иванка; Тернер, Эмили Х; Кумар, Акаш; Ховард, Элеонора; Шендюр, Джей; Тернер, Дэниел Дж; и др. (февраль 2010 г.). «Стратегии целевого обогащения для секвенирования следующего поколения». Природные методы . 7 (2): 111–118. дои : 10.1038/nmeth.1419 . ПМИД 20111037 . S2CID 21410733 .
- ^ Jump up to: а б с Бизекер Л.Г. (январь 2010 г.). «Секвенирование экзома делает медицинскую геномику реальностью» . Нат. Жене . 42 (1): 13–14. дои : 10.1038/ng0110-13 . ПМИД 20037612 .
- ^ Jump up to: а б Шварце, К; Бьюкенен, Дж; Тейлор, Дж. К.; Вордсворт, С. (май 2018 г.). «Являются ли подходы к секвенированию всего экзома и целого генома экономически эффективными? Систематический обзор литературы» . Ценность в здоровье . 21 : S100. дои : 10.1016/j.jval.2018.04.677 . ISSN 1098-3015 .
- ^ Ли, Сынгын; Абекасис, Гонсало Р.; Бенке, Майкл; Линь, Сихун (июль 2014 г.). «Анализ ассоциаций редких вариантов: планы исследования и статистические тесты» . Американский журнал генетики человека . 95 (1): 5–23. дои : 10.1016/j.ajhg.2014.06.009 . ПМЦ 4085641 . ПМИД 24995866 .
- ^ Ван, Цюаньли; Диндса, Райан С.; Карсс, Керен; Харпер, Эндрю Р.; и др. (23 сентября 2021 г.). «Вклад редких вариантов в болезни человека в 281 104 экзомах Британского биобанка» . Природа . 597 (7877): 527–532. Бибкод : 2021Natur.597..527W . дои : 10.1038/s41586-021-03855-y . ПМЦ 8458098 . ПМИД 34375979 .
- ^ Ли, Сихао; Ли, Жилин; Чжоу, Хуфэн; Гейнор, Шейла М.; и др. (сентябрь 2020 г.). «Динамическое включение нескольких функциональных аннотаций in silico расширяет возможности анализа ассоциаций редких вариантов в крупных масштабных исследованиях полногеномного секвенирования» . Природная генетика . 52 (9): 969–983. дои : 10.1038/s41588-020-0676-4 . ПМЦ 7483769 . ПМИД 32839606 .
- ^ «STAARpipeline: универсальный редкий вариант инструмента для данных полногеномного секвенирования в масштабе биобанка». Природные методы . 19 (12): 1532–1533. Декабрь 2022 г. doi : 10.1038/s41592-022-01641-w . ПМИД 36316564 . S2CID 253246835 .
- ^ Ли, Сихао; Быстрее, Корбин; Чжоу, Хуфэн; Гейнор, Шейла М.; Лю, Яову; Чен, Хан; Сельварадж, Маргарет Сунита; Сан, Райан; День, Рунак; Арнетт, Донна К.; Белак, Лоуренс Ф.; Бис, Джошуа К.; Бланджеро, Джон; Бурвинкль, Эрик; Боуден, Дональд В.; Броуди, Дженнифер А.; Кейд, Брайан Э.; Корреа, Адольфо; Капплс, Л. Эдриенн; Карран, Джоан Э.; де Врис, Поль С.; Дуггирала, Равиндранат; Фридман, Барри И.; Геринг, Харальд Х.Х.; Го, Сюцин; Хесслер, Джеффри; Кальяни, Рита Р.; Куперберг, Чарльз; Крал, Брайан Г.; Ланге, Лесли А.; Маничайкул, Ани; Мартин, Лиза В.; МакГарви, Стивен Т.; Митчелл, Брэкстон Д.; Монтассер, Мэй Э.; Моррисон, Аланна С.; Насери, Таке; О'Коннелл, Джеффри Р.; Палмер, Николетт Д.; Пейзер, Патрисия А.; Псати, Брюс М.; Раффилд, Лаура М.; Редлайн, Сьюзен; Райнер, Александр П.; Реупена, Муагутутиа Сефуива; Райс, Кеннет М.; Рич, Стивен С.; Ситлани, Коллин М.; Смит, Дженнифер А.; Тейлор, Кент Д.; Васан, Рамачандран С.; Уиллер, Кристен Дж.; Уилсон, Джеймс Г.; Янек, Лиза Р.; Чжао, Вэй; Консорциум NHLBI Trans-Omics for Precision Medicine (TOPMed); Рабочая группа TOPMed по липидам; Роттер, Джером И.; Натараджан, Прадип; Пелосо, Джина М.; Ли, Жилин; Линь, Сихун (январь 2023 г.). «Мощный, масштабируемый и ресурсоэффективный метаанализ ассоциаций редких вариантов в крупных исследованиях полногеномного секвенирования» . Природная генетика . 55 (1): 154–164. дои : 10.1038/s41588-022-01225-6 . ПМЦ 10084891 . ПМИД 36564505 . S2CID 255084231 .
- ^ Jump up to: а б Вэйцзюнь Ло; Чаолинь Чжан; Юн-хуэй Цзян; Кори Р. Брауэр (2018). «Систематическая реконструкция биологии аутизма на основе массивных профилей генетических мутаций» . Достижения науки . 4 (4): e1701799. Бибкод : 2018SciA....4.1799L . дои : 10.1126/sciadv.1701799 . ПМЦ 5895441 . ПМИД 29651456 .
- ^ Jump up to: а б Билгювар К., Озтюрк А.К., Луви А., Кван К.Ю., Чой М., Татли Б., Ялнизоглу Д., Тюйсюз Б., Чаглаян А.О., Гекбен С., Каймакчалан Х., Барак Т., Бакирчиоглу М., Ясуно К., Хо В, Сандерс С., Чжу Й. , Йылмаз С., Динчер А., Джонсон М.Х., Бронен Р.А., Кочер Н., Пер Х., Мане С., Памир М.Н., Ялчинкая С., Кумандаш С., Топчу М., Озмен М., Сестан Н., Лифтон Р.П., Стэйт М.В., Гюнель М. (9 сентября 2010 г.). «Секвенирование всего экзома выявляет рецессивные мутации WDR62 при тяжелых пороках развития головного мозга» . Природа . 467 (7312): 207–210. Бибкод : 2010Natur.467..207B . дои : 10.1038/nature09327 . ПМК 3129007 . ПМИД 20729831 .
- ^ Jump up to: а б с Уорти Э.А., Майер А.Н., Сиверсон Г.Д., Хелблинг Д., Боначчи Б.Б., Декер Б., Серпе Дж.М., Дасу Т., Чаннен М.Р., Вейт Р.Л., Бейсхор М.Дж., Брокель Ю., Томита-Митчелл А., Арка М.Дж., Каспер Дж.Т., Марголис Д.А., Бик Д.П., Хесснер М.Дж., Рутс Дж.М., Вербски Дж.В., Джейкоб Х.Дж., Диммок Д.П. (март 2011 г.). «Постановка окончательного диагноза: успешное клиническое применение секвенирования всего экзома у ребенка с трудноизлечимым воспалительным заболеванием кишечника» . Жене. Мед . 13 (3): 255–262. дои : 10.1097/GIM.0b013e3182088158 . ПМИД 21173700 .
- ^ Jump up to: а б Раффан Э., Херст Л.А., Турки С.А., Карпентер Г., Скотт С., Дейли А., Коффи А., Бхаскар С., Ховард Э., Хан Н., Кингстон Х., Палоти А., Сэвидж Д.Б., О’Дрисколл М., Смит С., О’Рахилли С., Баррозу И., Семпл Р.К. (2011). «Ранняя диагностика синдрома Вернера с использованием полноэкзомного секвенирования у одного атипичного пациента» . Фронт Эндокринол (Лозанна) . 2 (8): 8. дои : 10.3389/fendo.2011.00008 . ПМЦ 3356119 . ПМИД 22654791 .
- ^ Уорр, А.; Роберт, К.; Хьюм, Д.; Арчибальд, А.; Диб, Н.; Уотсон, М. (2 июля 2015 г.). «Секвенирование экзома: текущие и будущие перспективы» . Г3 . 5 (8): 1543–1550. дои : 10.1534/g3.115.018564 . ПМЦ 4528311 . ПМИД 26139844 .
- ^ Jump up to: а б Герпер, Мэтью (27 сентября 2011 г.). «Будущее уже сейчас: 23andMe теперь предлагает все ваши гены за 999 долларов» . Форбс . Проверено 11 декабря 2011 г.
- ^ «23andMe запускает пилотную программу прямого секвенирования экзома для потребителя» . ГеномВеб . ООО «ГеномВеб». 28 сентября 2011 года . Проверено 11 декабря 2011 г.
- ^ «Секвенирование личного генома у якобы здоровых людей и консорциума PeopleSeq» (PDF) . Геномы2Люди. 14 июня 2016 г.
- ^ Форхаус, Дэн (29 ноября 2012 г.). «DTC DTC: Возвращение прямого секвенирования генома к потребителям» . Отчет о законе о геномике . Проверено 30 мая 2013 г.
- ^ «Абсолютное продвижение Exome» . БГИ Америка. 18 октября 2013 года. Архивировано из оригинала 10 ноября 2013 года . Проверено 17 ноября 2013 г.
- ^ «Владение вашими данными: модель Genos» . Био-ИТ Мир. 6 июля 2016 г.
- ^ «Стартап Genos в области потребительской геномики планирует предоставить клиентам возможность исследовать и делиться своими данными» . ООО «ГеномВеб». 13 июня 2016 г.
- ^ «Генос: владей своей ДНК, изучай себя, проводи исследования» . www.genosresearch.com . Архивировано из оригинала 25 августа 2016 г. Проверено 18 октября 2016 г.