Чип-секвенирование
ChIP-секвенирование , также известное как ChIP-seq , представляет собой метод, используемый для анализа взаимодействий белка с ДНК . ChIP-seq сочетает в себе иммунопреципитацию хроматина (ChIP) с массово-параллельным секвенированием ДНК для идентификации сайтов связывания ДНК-ассоциированных белков. Его можно использовать для точного картирования глобальных сайтов связывания любого интересующего белка. Ранее ChIP-на-чипе был наиболее распространенным методом, используемым для изучения отношений белок-ДНК.
Использование
[ редактировать ]ChIP-seq в первую очередь используется для определения того, как факторы транскрипции и другие белки, ассоциированные с хроматином, влияют на механизмы, влияющие на фенотип . Определение того, как белки взаимодействуют с ДНК, регулируя экспрессию генов, имеет важное значение для полного понимания многих биологических процессов и болезненных состояний. Эта эпигенетическая информация дополняет анализ генотипа и экспрессии. Технология ChIP-seq в настоящее время рассматривается в первую очередь как альтернатива ChIP-чипу , требующему гибридизации . Это вносит некоторую предвзятость, поскольку массив ограничен фиксированным количеством зондов. Секвенирование, напротив, считается менее предвзятым, хотя смещение различных технологий секвенирования еще не до конца изучено. [1]
Специфические участки ДНК, находящиеся в прямом физическом взаимодействии с факторами транскрипции и другими белками, можно выделить методом иммунопреципитации хроматина . ChIP создает библиотеку целевых участков ДНК, связанных с интересующим белком. Массивно-параллельный анализ последовательностей используется в сочетании с базами данных последовательностей всего генома для анализа картины взаимодействия любого белка с ДНК. [2] или характер любых эпигенетических модификаций хроматина . Это можно применить к набору белков и модификаций, способных использовать ChIP, таких как факторы транскрипции, полимеразы и транскрипционные механизмы , структурные белки , модификации белков и модификации ДНК . [3] В качестве альтернативы зависимости от специфических антител были разработаны различные методы для поиска расширенного набора всех активных регуляторных областей с истощенными или разрушенными нуклеосомами в геноме, такие как DNase-Seq. [4] и FAIRE-Seq . [5] [6]
Рабочий процесс ChIP-секвенирования
[ редактировать ]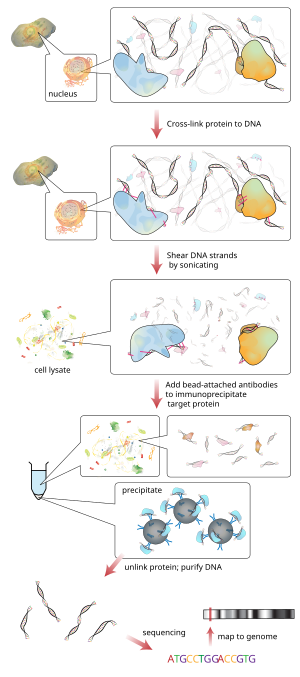
ЧИП
[ редактировать ]ChIP — это мощный метод избирательного обогащения последовательностей ДНК, связанных с определенным белком, в живых клетках . Однако широкое использование этого метода было ограничено отсутствием достаточно надежного метода для идентификации всех обогащенных последовательностей ДНК. Протокол влажной лаборатории ChIP содержит ChIP и гибридизацию. По сути, протокол ChIP состоит из пяти частей. [7] это помогает лучше понять весь процесс ChIP. Для проведения ЧИП первым шагом является перекрестное связывание. [8] используя формальдегид и большие партии ДНК, чтобы получить полезное количество. Поперечные связи образуются между белком и ДНК, а также между РНК и другими белками. Вторым шагом является процесс фрагментации хроматина, который разрушает хроматин, чтобы в конечном итоге получить высококачественные фрагменты ДНК для анализа ChIP. Эти фрагменты следует разрезать, чтобы их длина составляла менее 500 пар оснований. [9] каждый из них даст наилучший результат для картирования генома. Третий этап называется иммунопреципитацией хроматина. [7] это сокращение от ChIP. Процесс ChIP усиливает специфические сшитые комплексы ДНК-белок с использованием антитела против интересующего белка с последующей инкубацией и центрифугированием для получения иммунопреципитации. Стадия иммунопреципитации также позволяет удалить сайты неспецифического связывания. Четвертый шаг — восстановление и очистка ДНК. [7] происходящий путем обратного воздействия на сшивку между ДНК и белком с целью их разделения и очистки ДНК с помощью экстракции. Пятый и последний шаг — это этап анализа протокола ChIP с помощью процесса qPCR , ChIP-на-чипе (гибридный массив) или секвенирования ChIP. олигонуклеотидные Затем к небольшим участкам ДНК, которые были связаны с интересующим белком, добавляются адаптеры, чтобы обеспечить массовое параллельное секвенирование . Затем посредством анализа последовательности можно идентифицировать и интерпретировать по гену или участку, с которым был связан белок. [7]
Секвенирование
[ редактировать ]После выбора размера все полученные фрагменты ChIP-ДНК секвенируются одновременно с помощью секвенатора генома. За один цикл секвенирования можно сканировать общегеномные ассоциации с высоким разрешением, а это означает, что признаки могут быть расположены точно на хромосомах. Чип-чип, напротив, требует больших наборов тайловых массивов для меньшего разрешения. [10]
На этом этапе секвенирования используется множество новых методов секвенирования . Некоторые технологии, анализирующие последовательности, могут использовать кластерную амплификацию фрагментов ДНК ChIP, лигированных адаптером, на твердом субстрате проточных клеток для создания кластеров примерно по 1000 клональных копий каждый. Полученный массив шаблонных кластеров высокой плотности на поверхности проточной кюветы секвенируется с помощью программы анализа генома. Каждый матричный кластер подвергается секвенированию путем синтеза параллельно с использованием новых флуоресцентно меченных обратимых терминаторных нуклеотидов. Шаблоны секвенируются база за базой во время каждого чтения. Затем программное обеспечение для сбора и анализа данных выравнивает последовательности образцов с известной геномной последовательностью для идентификации фрагментов ChIP-ДНК. [ нужна ссылка ]
Контроль качества
[ редактировать ]ChIP-seq предлагает нам быстрый анализ, однако необходимо провести контроль качества, чтобы убедиться в достоверности полученных результатов:
- Неизбыточная фракция : области низкой сложности следует удалить, поскольку они неинформативны и могут мешать картированию в эталонном геноме. [11]
- Фрагменты в пиках: соотношение операций чтения, расположенных в пиках, к операциям чтения, расположенных там, где пика нет. [11]
Чувствительность
[ редактировать ]Чувствительность этой технологии зависит от глубины проведения секвенирования (т.е. количества картированных тегов последовательности), размера генома и распределения целевого фактора.Глубина секвенирования напрямую коррелирует со стоимостью. Если необходимо картировать большое количество связывающих веществ в больших геномах с высокой чувствительностью, затраты будут высокими, поскольку потребуется чрезвычайно большое количество меток последовательностей. В этом отличие от ЧИП-чипа, у которого стоимость не коррелирует с чувствительностью. [12] [13]
В отличие от методов ChIP на основе микрочипов , точность анализа ChIP-seq не ограничивается расстоянием между заранее определенными зондами. За счет интеграции большого количества коротких ридов достигается очень точная локализация сайта связывания. По сравнению с ChIP-чипом, данные ChIP-seq можно использовать для определения местоположения сайта связывания в пределах нескольких десятков пар оснований от фактического сайта связывания белка. Плотность меток в сайтах связывания является хорошим индикатором аффинности связывания белок-ДНК. [14] что облегчает количественную оценку и сравнение аффинности связывания белка с различными участками ДНК. [15]
Текущие исследования
[ редактировать ]Ассоциация ДНК STAT1: ChIP-seq использовали для изучения мишеней STAT1 в клетках HeLa S3, которые являются клонами линии HeLa и используются для анализа клеточных популяций. [16] Затем эффективность ChIP-seq сравнивали с альтернативными методами взаимодействия белок-ДНК, такими как ChIP-PCR и ChIP-чип. [17]
Нуклеосомная архитектура промоторов: с помощью ChIP-seq было установлено, что дрожжевые гены, по-видимому, имеют минимальную безнуклеосомную промоторную область длиной 150 п.н., в которой РНК-полимераза может инициировать транскрипцию. [18]
Консервация факторов транскрипции: ChIP-seq использовали для сравнения консервативности ТФ в тканях переднего мозга и сердца у эмбриональных мышей. Авторы идентифицировали и подтвердили функциональность энхансеров транскрипции в сердце и определили, что энхансеры транскрипции для сердца менее консервативны, чем энхансеры для переднего мозга на той же стадии развития. [19]
Полногеномное ChIP-секвенирование: ChIP-секвенирование было завершено на черве C. elegans для изучения полногеномных сайтов связывания 22 факторов транскрипции. До 20% аннотированных генов-кандидатов были отнесены к транскрипционным факторам. Несколько факторов транскрипции были отнесены к некодирующим областям РНК и могут зависеть от переменных развития или окружающей среды. Также были идентифицированы функции некоторых транскрипционных факторов. Некоторые из факторов транскрипции регулируют гены, которые контролируют другие факторы транскрипции. Эти гены не регулируются другими факторами. Большинство факторов транскрипции служат как мишенями, так и регуляторами других факторов, демонстрируя сеть регуляции. [20]
Предполагаемая регуляторная сеть: было показано, что сигнал ChIP-seq модификации гистонов в большей степени коррелирует с мотивами транскрипционных факторов на промоторах по сравнению с уровнем РНК. [21] Следовательно, автор предположил, что использование модификации гистонов ChIP-seq обеспечит более надежный вывод о генно-регуляторных сетях по сравнению с другими методами, основанными на экспрессии.
ChIP-seq предлагает альтернативу ChIP-чипу. Экспериментальные данные ChIP-seq STAT1 имеют высокую степень сходства с результатами, полученными с помощью ChIP-чипа для экспериментов того же типа, с более чем 64% пиков в общих геномных областях. Поскольку данные представляют собой считывание последовательностей, ChIP-seq предлагает конвейер быстрого анализа при условии, что высококачественная последовательность генома доступна для сопоставления чтения и в геноме нет повторяющегося содержимого, которое запутывает процесс сопоставления. ChIP-seq также потенциально может обнаруживать мутации в последовательностях сайтов связывания, что может напрямую поддерживать любые наблюдаемые изменения в связывании белков и регуляции генов.
Компьютерный анализ
[ редактировать ]Как и многие подходы к высокопроизводительному секвенированию, ChIP-seq генерирует чрезвычайно большие наборы данных, для которых требуются соответствующие методы компьютерного анализа. Для прогнозирования сайтов связывания ДНК на основе данных подсчета чтений ChIP-seq вызова пиков были разработаны методы . Один из самых популярных методов [ нужна ссылка ] - это MACS, который эмпирически моделирует размер сдвига тегов ChIP-Seq и использует его для улучшения пространственного разрешения прогнозируемых сайтов связывания. [22] MACS оптимизирован для пиков с более высоким разрешением, в то время как другой популярный алгоритм, SICER, запрограммирован на вызов более широких пиков, охватывающих от килобаз до мегабаз, для поиска более широких доменов хроматина. SICER более полезен для меток гистонов, охватывающих тела генов. Более строгий математический метод BCP (Bayesian Change Point) можно использовать как для острых, так и для широких пиков с более высокой скоростью вычислений. [23] см. сравнительное сравнение инструментов пикового вызова ChIP-seq, проведенное Thomas et al. (2017). [24]
Другой актуальной вычислительной проблемой является дифференциальный вызов пиков, который выявляет существенные различия в двух сигналах ChIP-seq из разных биологических условий. Вызывающие дифференциальные пики сегментируют два сигнала ChIP-seq и идентифицируют дифференциальные пики с помощью скрытых марковских моделей . Примерами двухэтапных дифференциальных пиковых вызывающих устройств являются ChIPDiff. [25] и ОДИН. [26]
Чтобы уменьшить количество ложных сайтов в результате ChIP-seq, можно использовать несколько экспериментальных контролей для обнаружения сайтов связывания в эксперименте по IP. Bay2Ctrls использует байесовскую модель для интеграции контроля ввода ДНК для IP, ложного IP и соответствующего контроля ввода ДНК для прогнозирования сайтов связывания на основе IP. [27] Этот подход особенно эффективен для сложных образцов, таких как целые модельные организмы. Кроме того, анализ показывает, что для сложных образцов контрольные образцы IP существенно превосходят контрольные образцы, вводимые ДНК, вероятно, из-за активных геномов образцов. [27]
См. также
[ редактировать ]Похожие методы
[ редактировать ]- Секвенирование CUT&RUN : контролируемое расщепление, направленное на антитела, с помощью микрококковой нуклеазы вместо ChIP, что позволяет улучшить соотношение сигнал/шум во время секвенирования.
- Секвенирование CUT&Tag , контролируемое расщепление, направленное на антитела, с помощью транспозазы Tn5 вместо ChIP, что позволяет улучшить соотношение сигнал/шум во время секвенирования.
- Sono-Seq , идентичен ChIP-Seq, но без этапа иммунопреципитации.
- ХИТ-КЛИП [28] [29] (также называемый CLIP-Seq ), для обнаружения взаимодействий с РНК, а не с ДНК.
- PAR-CLIP , еще один метод идентификации сайтов связывания клеточных РНК-связывающих белков (RBP).
- RIP-Chip , та же цель и первые шаги, но не использует методы перекрестного связывания и использует микроматрицы вместо секвенирования.
- SELEX — метод поиска консенсусной связывающей последовательности.
- Конкуренция-ЧИП , для измерения динамики относительной замены ДНК.
- ChiRP-Seq для измерения РНК-связанной ДНК и белков.
- ChIP-exo использует обработку экзонуклеазой для достижения разрешения до одной пары оснований.
- ChIP-nexus улучшенная версия ChIP-exo для достижения разрешения до одной пары оснований.
- DRIP-seq использует антитело S9.6 для преципитации трехцепочечных гибридов DND:РНК, называемых R-петлями.
- TCP-seq , принципиально аналогичный метод измерения динамики трансляции мРНК.
- «Визитные карты» используют транспозазу для обозначения последовательности, в которой связывается фактор транскрипции. [30]
Ссылки
[ редактировать ]- ^ Мухаммад, Исиака Ибрагим; Конг, Сзе Лин; Акмар Абдулла, Сити Нор; Мунусами, Умайял (25 декабря 2019 г.). «РНК-сек и ChIP-сек как дополнительные подходы к пониманию механизма регуляции транскрипции растений» . Международный журнал молекулярных наук . 21 (1): 167. doi : 10.3390/ijms21010167 . ISSN 1422-0067 . ПМК 6981605 . ПМИД 31881735 .
- ^ Джонсон Д.С., Мортазави А., Майерс Р.М., Уолд Б. (июнь 2007 г.). «Полногеномное картирование взаимодействий белка и ДНК in vivo» (PDF) . Наука . 316 (5830): 1497–502. Бибкод : 2007Sci...316.1497J . дои : 10.1126/science.1141319 . ПМИД 17540862 . S2CID 519841 .
- ^ «Полногеномное IP-секвенирование хроматина (ChIP-Seq)» (PDF) . Illumina, Inc., 26 ноября 2007 г.
- ^ Сун, Линюнь; Кроуфорд, Грегори Э. (февраль 2010 г.). «DNase-seq: метод высокого разрешения для картирования активных генных регуляторных элементов по всему геному клеток млекопитающих» . Протоколы Колд-Спринг-Харбора . 2010 (2): pdb.prot5384. дои : 10.1101/pdb.prot5384 . ISSN 1559-6095 . ПМЦ 3627383 . ПМИД 20150147 .
- ^ Гирези, Пол Г.; Ким, Джонхван; МакДэниел, Райан М.; Айер, Вишванат Р.; Либ, Джейсон Д. (июнь 2007 г.). «FAIRE (выделение регуляторных элементов с помощью формальдегида) изолирует активные регуляторные элементы из хроматина человека» . Геномные исследования . 17 (6): 877–885. дои : 10.1101/гр.5533506 . ISSN 1088-9051 . ЧВК 1891346 . ПМИД 17179217 .
- ^ Кумар, Вибхор; Муратани, Масафуми; Райан, Нирмала Арул; Краус, Петра; Лафкин, Томас; Нг, Хак Хуэй; Прабхакар, Шьям (июль 2013 г.). «Единая, оптимальная обработка сигналов сопоставленных данных глубокого секвенирования» . Природная биотехнология . 31 (7): 615–622. дои : 10.1038/nbt.2596 . ISSN 1087-0156 . ПМИД 23770639 . S2CID 32510475 .
- ^ Перейти обратно: а б с д «Руководство по ChIP: приложения эпигенетики | Abcam» . www.abcam.com . Проверено 2 марта 2020 г.
- ^ Ким Т.Х., Деккер Дж. (апрель 2018 г.). «Формальдегидная сшивка». Протоколы Колд-Спринг-Харбора . 2018 (4): pdb.prot082594. дои : 10.1101/pdb.prot082594 . ПМИД 29610357 .
- ^ Ким Т.Х., Деккер Дж. (апрель 2018 г.). «Подготовка сшитого хроматина для ЧИП». Протоколы Колд-Спринг-Харбора . 2018 (4): pdb.prot082602. дои : 10.1101/pdb.prot082602 . ПМИД 29610358 .
- ^ Парк, Питер Дж. (октябрь 2009 г.). «ЧИП-сек: преимущества и проблемы развивающейся технологии» . Обзоры природы. Генетика . 10 (10): 669–680. дои : 10.1038/nrg2641 . ISSN 1471-0064 . ПМК 3191340 . ПМИД 19736561 .
- ^ Перейти обратно: а б Чен, Ивэнь; Негре, Николя; Ли, Цюньхуа; Мечковска, Джоанна О.; Слэттери, Мэтью; Лю, Тао; Чжан, Юн; Ким, Тэ Гён; Он, Хоушенг Хансен; Зиеба, Дженнифер; Жуан, Иджун (июнь 2012 г.). «Систематическая оценка факторов, влияющих на точность ChIP-seq» . Природные методы . 9 (6): 609–614. doi : 10.1038/nmeth.1985 . ISSN 1548-7105 . ПМЦ 3477507 . ПМИД 22522655 .
- ^ Юнг, Янгсук Л.; Люкетт, Лавлейс Дж.; Хо, Джошуа В.К.; Феррари, Франческо; Толсторуков, Михаил; Минода, Аки; Исснер, Роббин; Эпштейн, Чарльз Б.; Карпен, Гэри Х.; Курода, Митци И.; Парк, Питер Дж. (май 2014 г.). «Влияние глубины секвенирования в экспериментах ChIP-seq» . Исследования нуклеиновых кислот . 42 (9): е74. дои : 10.1093/nar/gku178 . ISSN 1362-4962 . ПМК 4027199 . ПМИД 24598259 .
- ^ Хо, Джошуа В.К.; Бишоп, Эрик; Карченко Петр Владимирович; Негре, Николя; Уайт, Кевин П.; Парк, Питер Дж. (28 февраля 2011 г.). «ChIP-чип против ChIP-seq: уроки планирования экспериментов и анализа данных» . БМК Геномика . 12 :134. дои : 10.1186/1471-2164-12-134 . ISSN 1471-2164 . ПМК 3053263 . ПМИД 21356108 .
- ^ Джоти и др. (2008). «Полногеномная идентификация сайтов связывания белка с ДНК in vivo на основе данных ChIP-seq» . Нуклеиновые кислоты Рез . 36 (16): 5221–5231. дои : 10.1093/нар/gkn488 . ПМЦ 2532738 . ПМИД 18684996 .
- ^ Бернштейн Б.Е., Камаль М., Линдблад-Тох К., Бекиранов С., Бэйли Д.К., Хьюберт Д.Д. и др. (январь 2005 г.). «Геномные карты и сравнительный анализ модификаций гистонов у человека и мыши» . Клетка . 120 (2): 169–81. дои : 10.1016/j.cell.2005.01.001 . ПМИД 15680324 . S2CID 7193829 .
- ^ «HeLa S3 от ATCC | Biocompare.com» . www.biocompare.com . Проверено 21 марта 2020 г.
- ^ Робертсон Г., Херст М., Бейнбридж М., Биленки М., Чжао Ю., Цзэн Т. и др. (август 2007 г.). «Пологеномные профили ассоциации ДНК STAT1 с использованием иммунопреципитации хроматина и массового параллельного секвенирования». Природные методы . 4 (8): 651–7. дои : 10.1038/nmeth1068 . ПМИД 17558387 . S2CID 28531263 .
- ^ Шмид CD, Бухер П. (ноябрь 2007 г.). «Данные ChIP-Seq раскрывают нуклеосомную архитектуру человеческих промоторов» . Клетка . 131 (5): 831–2, ответ автора 832–3. дои : 10.1016/j.cell.2007.11.017 . ПМИД 18045524 . S2CID 29234049 .
- ^ Блоу MJ, Маккалли DJ, Ли Зи, Чжан Т, Акияма Дж.А., Холт А. и др. (сентябрь 2010 г.). «ChIP-Seq идентификация слабоконсервативных усилителей сердца» . Природная генетика . 42 (9): 806–10. дои : 10.1038/ng.650 . ПМК 3138496 . ПМИД 20729851 .
- ^ Ню В., Лу З.Дж., Чжун М., Саров М., Мюррей Дж.И., Брдлик С.М. и др. (февраль 2011 г.). «Различные особенности связывания факторов транскрипции, выявленные с помощью полногеномной ChIP-секвенации у C. elegans» . Геномные исследования . 21 (2): 245–54. дои : 10.1101/гр.114587.110 . ПМК 3032928 . ПМИД 21177963 .
- ^ Кумар В., Муратани М., Райан Н.А., Краус П., Луфкин Т., Нг Х.Х., Прабхакар С. (июль 2013 г.). «Единая, оптимальная обработка сигналов сопоставленных данных глубокого секвенирования» . Природная биотехнология . 31 (7): 615–22. дои : 10.1038/nbt.2596 . ПМИД 23770639 .
- ^ Чжан Ю., Лю Т., Мейер К.А., Экхаут Дж., Джонсон Д.С., Бернштейн Б.Е. и др. (2008). «Модельный анализ ChIP-Seq (MACS)» . Геномная биология . 9 (9): 137 р. дои : 10.1186/gb-2008-9-9-r137 . ПМК 2592715 . ПМИД 18798982 .
- ^ Син Х, Мо Ю, Ляо В, Чжан МЦ (2012). «Пологеномная локализация связывания белок-ДНК и модификация гистонов с помощью байесовского метода точки изменения с данными ChIP-seq» . ПЛОС Компьютерная Биол . 8 (7): e1002613. Бибкод : 2012PLSCB...8E2613X . дои : 10.1371/journal.pcbi.1002613 . ПМК 5429005 . ПМИД 22844240 .
- ^ Томас Р., Томас С., Холлоуэй А.К., Поллард К.С. (2017). «Функции, определяющие лучшие алгоритмы пикового вызова ChIP-seq» . Краткий Биоинформ . 18 (3): 441–450. дои : 10.1093/нагрудник/bbw035 . ПМК 5429005 . ПМИД 27169896 .
- ^ Сюй Х, Вэй С.Л., Линь Ф., Сун В.К. (октябрь 2008 г.). «Подход HMM к полногеномной идентификации сайтов дифференциальной модификации гистонов на основе данных ChIP-seq» . Биоинформатика . 24 (20): 2344–9. doi : 10.1093/биоинформатика/btn402 . ПМИД 18667444 .
- ^ Аллхофф М., Сере К., Шовистре Х., Лин К., Зенке М., Коста И.Г. (декабрь 2014 г.). «Обнаружение дифференциальных пиков в сигналах ChIP-seq с помощью ODIN». Биоинформатика . 30 (24): 3467–75. doi : 10.1093/биоинформатика/btu722 . ПМИД 25371479 .
- ^ Перейти обратно: а б Сюй, Цзиньруй; Кудрон, Мишель М; Викторсен, Алек; Гао, Цзяхао; Аммури, Ханин Н; Наварро, Фабио КП; Гевирцман, Луи; Уотерстон, Роберт Х; Уайт, Кевин П; Рейнке, Валери; Герштейн, Марк (21 декабря 2020 г.). «Издеваться или нет: всестороннее сравнение ложного IP и ввода ДНК для ChIP-seq» . Исследования нуклеиновых кислот . 49 (3): gkaa1155. дои : 10.1093/nar/gkaa1155 . ISSN 0305-1048 . ПМЦ 7897498 . ПМИД 33347581 .
- ^ Ликаталоси Д.Д., Меле А., Фак Дж.Дж., Уле Дж., Кайикчи М., Чи С.В. и др. (ноябрь 2008 г.). «HITS-CLIP дает общегеномное представление об альтернативной обработке РНК в мозге» . Природа . 456 (7221): 464–9. Бибкод : 2008Natur.456..464L . дои : 10.1038/nature07488 . ПМК 2597294 . ПМИД 18978773 .
- ^ Дарнелл РБ (2010). «HITS-CLIP: панорамный обзор регуляции белок-РНК в живых клетках» . Междисциплинарные обзоры Wiley. РНК . 1 (2): 266–286. дои : 10.1002/wrna.31 . ПМЦ 3222227 . ПМИД 21935890 .
- ^ Ван Х, Мэйхью Д., Чен X, Джонстон М., Митра Р.Д. (май 2011 г.). «Визитные карточки позволяют мультиплексную идентификацию геномных мишеней ДНК-связывающих белков» . Геномные исследования . 21 (5): 748–55. дои : 10.1101/гр.114850.110 . ПМК 3083092 . ПМИД 21471402 .
Внешние ссылки
[ редактировать ]- Каталог ReMap : интегративный и единый анализ регуляторных элементов ChIP-Seq из более чем 2800 наборов данных ChIP-seq, дающий каталог из 80 миллионов пиков от 485 регуляторов транскрипции. [1]
- База данных ChIPBase : база данных для изучения карт связывания факторов транскрипции на основе данных ChIP-Seq . Он предоставляет наиболее полный набор данных ChIP-Seq для различных типов клеток/тканей и состояний.
- База данных GeneProf и инструмент анализа : GeneProf — это свободно доступная и простая в использовании среда анализа данных ChIP-seq и RNA-seq, которая поставляется с большой базой данных готовых к анализу общедоступных экспериментов, например, по связыванию транскрипционных факторов и модификациям гистонов.
- Вызов дифференциального пика. Архивировано 15 мая 2021 г. на Wayback Machine : Учебное пособие по вызову дифференциального пика с помощью ODIN.
- Биоинформатический анализ данных ChIP-seq : Комплексный анализ данных ChIP-seq. [2]
- KLTepigenome : выявление коррелированной изменчивости в наборах эпигеномных данных с использованием преобразования Карунена-Лёве.
- SignalSpider : инструмент для обнаружения вероятностных закономерностей в нескольких нормализованных ChIP-Seq. профилях сигналов
- FullSignalRanker : инструмент для регрессии и прогнозирования пиков по множеству нормализованных ChIP-Seq. профилей сигналов
- ^ Ченеби Дж., Георг М., Артуфель М., Мателье А., Баллестер Б. (январь 2018 г.). «ReMap 2018: обновленный атлас регуляторных областей на основе интегративного анализа ДНК-связывающих экспериментов ChIP-seq» . Исследования нуклеиновых кислот . 46 (Д1): Д267–Д275. дои : 10.1093/нар/gkx1092 . ПМЦ 5753247 . ПМИД 29126285 .
- ^ Бэйли Т., Краевски П., Ладунга И., Лефевр С., Ли Кью, Лю Т. и др. (2013). «Практические рекомендации по комплексному анализу данных ChIP-seq» . PLOS Вычислительная биология . 9 (11): e1003326. Бибкод : 2013PLSCB...9E3326B . дои : 10.1371/journal.pcbi.1003326 . ПМЦ 3828144 . ПМИД 24244136 .