Модель замещения
В биологии модель замещения , также называемая моделями эволюции последовательности , представляет собой марковские модели , которые описывают изменения с течением эволюционного времени. Эти модели описывают эволюционные изменения в макромолекулах, таких как последовательности ДНК или последовательности белков , которые можно представить в виде последовательности символов (например, A, C, G и T в случае ДНК или 20 «стандартных» протеиногенных аминокислот в случае ДНК). случае белков ). Модели замещения используются для расчета вероятности с филогенетических деревьев использованием данных множественного выравнивания последовательностей . Таким образом, модели замещения играют центральную роль в оценке максимального правдоподобия филогении, а также в байесовском выводе в филогении . Оценки эволюционных расстояний (количества замен, произошедших с тех пор, как пара последовательностей отделилась от общего предка) обычно рассчитываются с использованием моделей замещения (эволюционные расстояния используются в качестве входных данных для методов расстояний, таких как присоединение соседей ). Модели замещения также играют центральную роль в филогенетических инвариантах , поскольку они необходимы для прогнозирования частот шаблонов сайтов с учетом древовидной топологии. Модели замещения также необходимы для моделирования данных о последовательностях для группы организмов, связанных определенным деревом.
Топологии филогенетического дерева и другие параметры
[ редактировать ]Топологии филогенетического дерева часто представляют интерес; [1] таким образом, длины ветвей и любые другие параметры, описывающие процесс замещения, часто рассматриваются как мешающие параметры . Однако биологов иногда интересуют и другие аспекты модели. Например, длины ветвей, особенно когда эти длины ветвей сочетаются с информацией из летописи окаменелостей и моделью для оценки временных рамок эволюции. [2] Другие параметры модели использовались для понимания различных аспектов процесса эволюции. Отношение K a /K s (также называемое ω в моделях замены кодонов) представляет собой параметр, представляющий интерес во многих исследованиях. Соотношение K a /K s можно использовать для изучения действия естественного отбора на области, кодирующие белки. [3] он предоставляет информацию об относительной частоте замен нуклеотидов, которые заменяют аминокислоты (несинонимичные замены), по сравнению с теми, которые не меняют кодируемую аминокислоту (синонимические замены).
Применение к данным секвенирования
[ редактировать ]Большая часть работ по моделям замещения была сосредоточена на эволюции последовательностей ДНК/ РНК и белков . Модели эволюции последовательности ДНК, в которых алфавит соответствует четырем нуклеотидам (A, C, G и T), вероятно, являются самыми простыми для понимания моделями. Модели ДНК также можно использовать для изучения эволюции РНК-вируса ; это отражает тот факт, что РНК также имеет четырехнуклеотидный алфавит (A, C, G и U). Однако модели замены можно использовать для алфавитов любого размера; Алфавит представляет собой 20 протеиногенных аминокислот для белков и смысловые кодоны (т.е. 61 кодон, которые кодируют аминокислоты в стандартном генетическом коде ) для выровненных последовательностей генов, кодирующих белок. Фактически, модели замещения могут быть разработаны для любых биологических признаков, которые можно закодировать с использованием определенного алфавита (например, аминокислотные последовательности в сочетании с информацией о конформации этих аминокислот в трехмерных белковых структурах). [4] ).
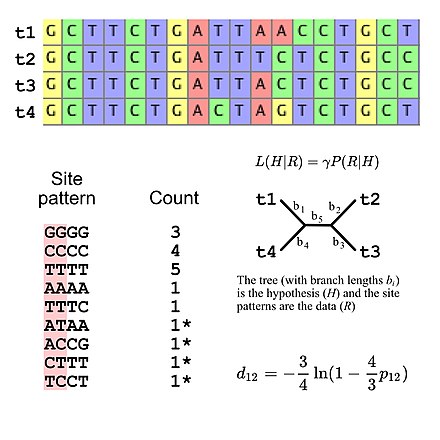
Большинство моделей замещения, используемых в эволюционных исследованиях, предполагают независимость сайтов (т.е. вероятность наблюдения любого конкретного шаблона сайтов идентична независимо от того, где шаблон сайтов находится в выравнивании последовательностей). Это упрощает расчеты вероятности, поскольку необходимо только вычислить вероятность всех шаблонов сайтов, которые появляются в выравнивании, а затем использовать эти значения для расчета общей вероятности совпадения (например, вероятность трех шаблонов сайтов «GGGG» с учетом некоторой модели Эволюция последовательности ДНК — это просто вероятность существования одного шаблона сайта «GGGG», возведенная в третью степень). Это означает, что модели замещения можно рассматривать как предполагающие определенное полиномиальное распределение частот шаблонов сайтов. Если мы рассмотрим множественное выравнивание четырех последовательностей ДНК, то существует 256 возможных шаблонов сайтов, то есть существует 255 степеней свободы для частот шаблонов сайтов. Однако можно указать ожидаемые частоты шаблонов сайтов, используя пять степеней свободы, если использовать модель эволюции ДНК Джукса-Кантора. [5] это простая модель замещения, которая позволяет рассчитать ожидаемые частоты шаблонов сайтов только топологию дерева и длину ветвей (для четырех таксонов некорневое разветвляющееся дерево имеет пять длин ветвей).
Модели замещения также позволяют моделировать данные о последовательностях с использованием методов Монте-Карло . Смоделированное множественное выравнивание последовательностей можно использовать для оценки эффективности филогенетических методов. [6] и генерировать нулевое распределение для некоторых статистических тестов в области молекулярной эволюции и молекулярной филогенетики. Примеры этих тестов включают тесты на соответствие модели. [7] и «тест SOWH», который можно использовать для проверки древовидных топологий. [8] [9]
Приложение к морфологическим данным
[ редактировать ]Тот факт, что модели замещения можно использовать для анализа любого биологического алфавита, позволил разработать модели эволюции для наборов фенотипических данных. [10] (например, морфологические и поведенческие особенности). Обычно это «0». используется для обозначения отсутствия черты, а «1» используется для обозначения наличия черты, хотя также возможно оценивать персонажей, используя несколько состояний. Используя эту структуру, мы могли бы закодировать набор фенотипов в виде двоичных строк (это можно обобщить до строк с k -состоянием для символов с более чем двумя состояниями) перед анализом с использованием соответствующего режима. Это можно проиллюстрировать на «игрушечном» примере: мы можем использовать двоичный алфавит для оценки следующих фенотипических признаков: «имеет перья», «откладывает яйца», «имеет мех», «теплокровен» и «способен к электропитанию». полет". В этом игрушечном примере у колибри будет последовательность 11011 (большинство других птиц будет иметь такую же последовательность), у страусов будет последовательность 11010, у крупного рогатого скота (и большинства других наземных млекопитающих ) будет 00110, а у летучих мышей будет 00111. Вероятность филогенетического Затем дерево можно рассчитать с использованием этих двоичных последовательностей и соответствующей модели замещения. Существование этих морфологических моделей позволяет анализировать матрицы данных с ископаемыми таксонами, используя только морфологические данные. [11] или сочетание морфологических и молекулярных данных [12] (последние считаются отсутствующими данными для ископаемых таксонов).
Существует очевидное сходство между использованием молекулярных или фенотипических данных в области кладистики и анализом морфологических признаков с использованием модели замещения. Однако разгорелась ожесточенная дискуссия [а] в сообществе систематиков относительно вопроса о том, следует ли рассматривать кладистический анализ как «свободный от моделей». Область кладистики (определенная в самом строгом смысле) предпочитает использование критерия максимальной экономии для филогенетических выводов. [13] Многие кладисты отвергают позицию, согласно которой максимальная экономия основана на модели замещения, и (во многих случаях) оправдывают использование экономности, используя философию Карла Поппера . [14] Однако существование «экономных эквивалентных» моделей [15] (т. е. модели замещения, которые при использовании для анализа дают дерево максимальной экономии) позволяют рассматривать экономию как модель замещения. [1]
Молекулярные часы и единицы времени
[ редактировать ]Обычно длина ветви филогенетического дерева выражается как ожидаемое количество замен на сайт; если эволюционная модель указывает, что каждый участок в предковой последовательности обычно будет претерпевать x замен к тому времени, когда он эволюционирует до определенной последовательности потомка, тогда считается, что предок и потомок разделены длиной ветви x .
Иногда длину ветви измеряют в геологических годах. Например, летопись окаменелостей может позволить определить количество лет между видом-предком и видом-потомком. Поскольку некоторые виды развиваются быстрее, чем другие, эти два показателя длины ветвей не всегда находятся в прямой зависимости. Ожидаемое количество замен на сайт в год часто обозначается греческой буквой мю (μ).
Говорят, что модель имеет строгие молекулярные часы , если ожидаемое количество замен в год μ постоянно, независимо от того, эволюцию какого вида исследуют. Важным следствием строгих молекулярных часов является то, что количество ожидаемых замен между предковым видом и любым из его современных потомков должно быть независимым от того, какой вид-потомок исследуется.
Обратите внимание, что предположение о строгих молекулярных часах часто нереалистично, особенно на длительных периодах эволюции. Например, хотя грызуны генетически очень похожи на приматов , они претерпели гораздо большее количество замен за предполагаемое время с момента дивергенции в некоторых областях генома . [16] Это может быть связано с более коротким временем генерации . [17] более высокая скорость метаболизма , усиление структурирования популяции, увеличение скорости видообразования или меньший размер тела . [18] [19] При изучении древних событий, таких как кембрийский взрыв , с использованием предположения о молекулярных часах, плохое совпадение между кладистическими часто наблюдается и филогенетическими данными. Была проведена некоторая работа над моделями, допускающими переменную скорость эволюции. [20] [21]
Модели, которые могут учитывать изменчивость хода молекулярных часов между разными эволюционными линиями филогенеза, называются «расслабленными» в отличие от «строгих». В таких моделях можно предположить, что уровень коррелирует или нет между предками и потомками, а вариация уровня между линиями может быть получена из многих распределений, но обычно применяются экспоненциальное и логнормальное распределения. Существует особый случай, называемый «локальными молекулярными часами», когда филогения разделена как минимум на два раздела (набора линий) и в каждом применяются строгие молекулярные часы, но с разной скоростью.
Обратимые во времени и стационарные модели
[ редактировать ]Многие полезные модели замещения обратимы во времени ; с точки зрения математики, модель не заботится о том, какая последовательность является предком, а какая — потомком, пока все остальные параметры (например, количество замен на сайт, ожидаемое между двумя последовательностями) остаются постоянными.
Когда проводится анализ реальных биологических данных, обычно нет доступа к последовательностям предковых видов, а только к современным видам. Однако если модель обратима во времени, то, какой вид был предковым видом, не имеет значения. Вместо этого филогенетическое дерево может быть укоренено с использованием любого из видов, повторно укоренено позже на основе новых знаний или оставлено неукорененным. Это потому, что не существует «особых» видов: все виды в конечном итоге произошли друг от друга с одинаковой вероятностью.
Модель обратима во времени тогда и только тогда, когда она удовлетворяет свойству (обозначения поясняются ниже)

или, что то же самое, свойство подробного баланса ,

для каждого i , j и t .
Обратимость во времени не следует путать со стационарностью . Модель является стационарной, если Q не меняется со временем. Приведенный ниже анализ предполагает стационарную модель.
Математика моделей замещения
[ редактировать ]Стационарные, нейтральные, независимые модели с конечными узлами (при условии постоянной скорости эволюции) имеют два параметра: π , равновесный вектор базовых (или характерных) частот и матрицу скоростей Q , которая описывает скорость, с которой базы одного типа переодеться в базы другого типа; элемент for i ≠ j — это скорость, с которой база i переходит в базу j . Диагонали матрицы Q выбираются так, чтобы сумма строк была равна нулю:


Равновесный вектор-строка π должен быть аннулирован матрицей скоростей Q :

Матрица-функция перехода представляет собой функцию от длин ветвей (в некоторых единицах времени, возможно, в подстановках) к матрице условных вероятностей. Он обозначается . Запись в я й столбец и j й ряд, , - это вероятность того, что после времени t существует база j в данной позиции, при условии, что база i находится в этой позиции в момент времени 0. Когда модель обратима во времени, это может быть выполнено между любыми двумя последовательностями, даже если один не является предком другого, если вы знаете общую длину ветвей между ними.


Асимптотические свойства P ij (t) таковы, что P ij (0) = δ ij , где δ ij — дельта -функция Кронекера. То есть нет никаких изменений в базовом составе между последовательностью и самой собой. В другой крайности, или, другими словами, когда время стремится к бесконечности, вероятность найти базу j в позиции, при условии, что в этой позиции была база i, первоначально превращается в равновесную вероятность того, что в этой позиции есть база j , независимо от исходной базы. Кроме того, отсюда следует, что для всех т .


Матрица перехода может быть вычислена из матрицы скоростей посредством возведения матрицы в степень :

где Q н матрица Q умножена сама на себя достаточное количество раз, чтобы получить ее n й власть.
Если Q диагонализуемо = , матричную экспоненту можно напрямую : пусть Q вычислить U −1 Λ U — диагонализация Q , причем

где Λ — диагональная матрица и где являются собственными значениями Q , каждое из которых повторяется в соответствии со своей кратностью. Затем


где диагональная матрица e Λt дается

Обобщенный обратимый во времени
[ редактировать ]Обобщенная обратимая во времени (GTR) - это наиболее общая нейтральная, независимая, обратимая во времени модель с конечными узлами. Впервые в общей форме он был описан Симоном Таваре в 1986 году. [22] Модель ОТО в публикациях часто называют общей обратимой во времени моделью; [23] ее также называют моделью REV. [24]
Параметры GTR для нуклеотидов состоят из равновесного вектора базовой частоты, , определяющий частоту, с которой каждое основание встречается на каждом сайте, и матрицу скоростей


Поскольку модель должна быть обратимой во времени и должна приближаться к равновесным частотам нуклеотидов (оснований) в течение длительного времени, каждая скорость ниже диагонали равна обратной скорости выше диагонали, умноженной на равновесное соотношение двух оснований. Таким образом, нуклеотидный GTR требует 6 параметров скорости замещения и 4 параметров равновесной базовой частоты. Поскольку сумма четырех частотных параметров должна быть равна 1, существует только три свободных частотных параметра. Общее количество свободных параметров часто сокращается до 8 плюс , общее количество замен в единицу времени. При измерении времени в заменах ( =1) осталось всего 8 свободных параметров.

Как правило, чтобы вычислить количество параметров, вы подсчитываете количество записей над диагональю в матрице, т. е. для n значений признаков на сайт. , а затем прибавьте n-1 для равновесных частот и вычтите 1, потому что фиксировано. Вы получаете


Например, для аминокислотной последовательности (есть 20 «стандартных» аминокислот , составляющих белки ) вы обнаружите 208 параметров. Однако при изучении кодирующих участков генома чаще работают с моделью замены кодонов (кодон состоит из трех оснований и кодирует одну аминокислоту в белке). Есть кодонов, что дает 2078 свободных параметров. Однако скорости переходов между кодонами, которые различаются более чем на одно основание, часто считаются равными нулю, что сокращает количество свободных параметров до всего лишь параметры. Другой распространенной практикой является уменьшение количества кодонов путем запрета стоп-кодонов (или бессмысленных кодонов). Это биологически разумное предположение, поскольку включение стоп-кодонов означало бы расчет вероятности обнаружения смыслового кодона. спустя время учитывая, что предковый кодон будет включать в себя возможность прохождения состояния с преждевременным стоп-кодоном.





Альтернативный (и часто используемый) [23] [25] [26] [27] ) способ записи матрицы мгновенных скоростей ( матрица) для нуклеотидной модели GTR:


The матрица нормализована, поэтому .

Это обозначение легче понять, чем обозначение, первоначально использованное Таваре , поскольку все параметры модели соответствуют либо параметрам «заменяемости» ( через , что также можно записать, используя обозначения ) или к равновесным нуклеотидов частотам . Обратите внимание, что нуклеотиды в матрицы записаны в алфавитном порядке. Другими словами, матрица вероятности перехода для матрица выше будет:





В некоторых публикациях нуклеотиды пишутся в другом порядке (например, некоторые авторы предпочитают группировать два пурина вместе и два пиримидина вместе; см. также модели эволюции ДНК ). Эти различия в обозначениях делают важным четко понимать порядок состояний при написании матрица.
Ценность этого обозначения состоит в том, что мгновенная скорость изменения нуклеотида в нуклеотид всегда можно записать как , где это обменность нуклеотидов и и равновесная частота нуклеотид. В приведенной выше матрице используются буквы через для параметров взаимозаменяемости в интересах удобства чтения, но эти параметры также можно записать систематическим образом, используя обозначение (например, , и так далее).





Обратите внимание, что порядок нуклеотидных индексов для параметров обменности не имеет значения (например, ), но значения матрицы вероятностей перехода — нет (т. е. — это вероятность наблюдения A в последовательности 1 и C в последовательности 2, когда эволюционное расстояние между этими последовательностями равно тогда как — вероятность наблюдения C в последовательности 1 и A в последовательности 2 на одном и том же эволюционном расстоянии).



Произвольно выбранные параметры обменности (например, ) обычно устанавливается на значение 1, чтобы повысить читаемость оценок параметра обменности (поскольку он позволяет пользователям выражать эти значения относительно выбранного параметра обменности). Практика выражения параметров обменности в относительных величинах не представляет проблем, поскольку матрица нормализована. Нормализация позволяет (время) в матричном возведении в степень быть выражено в единицах ожидаемых замен на сайт (стандартная практика молекулярной филогенетики). Это эквивалентно утверждению, что устанавливается частота мутаций. до 1) и уменьшив количество свободных параметров до восьми. В частности, существует пять свободных параметров заменяемости ( через , которые выражены относительно фиксированного в этом примере) и три параметра равновесной базовой частоты (как описано выше, только три значения необходимо указать, потому что должна в сумме равняться 1).






Альтернативные обозначения также облегчают понимание подмоделей модели GTR, которые просто соответствуют случаям, когда параметры взаимозаменяемости и/или равновесной базовой частоты ограничены принятием равных значений. Был назван ряд конкретных подмоделей, в основном на основе их оригинальных публикаций:
| Модель | Параметры взаимозаменяемости | Параметры базовой частоты | Ссылка |
|---|---|---|---|
| JC69 (или JC) | Джукс и Кантор (1969) [5] | ||
| F81 | все ценности свободны | Фельзенштейн (1981) [28] | |
| К2П (или К80) | ( трансверсии ), ( переходы ) | Кимура (1980) [29] | |
| HKY85 | ( трансверсии ), ( переходы ) | все ценности свободны | Хасегава и др. (1985) [30] |
| К3СТ (или К81) | ( трансверсии ), ( трансверсии ), ( переходы ) | Кимура (1981) [31] | |
| ТН93 | ( трансверсии ), ( переходы ), ( переходы ) | все ценности свободны | Тамура и Ней (1993) [32] |
| СИМ | все параметры взаимозаменяемости бесплатно | Zharkikh (1994) [33] | |
| ГТР (или РЕД. [24] ) | все параметры взаимозаменяемости бесплатно | все ценности свободны | Таваре (1986) [22] |











Существует 203 возможных способа ограничения параметров взаимозаменяемости для формирования подмоделей ОТО. [34] начиная с JC69 [5] и F81 [28] модели (где все параметры взаимозаменяемости равны) на SYM [33] модель и полный GTR [22] (или РЕД. [24] ) модель (где все параметры взаимозаменяемости свободны). Равновесные базовые частоты обычно рассматриваются двумя разными способами: 1) все значения ограничены равными (т.е. ); или 2) все значения рассматриваются как свободные параметры. Хотя равновесные базовые частоты можно ограничить и другими способами, большинство ограничений связывают некоторые, но не все ценности нереалистичны с биологической точки зрения. Возможным исключением является соблюдение симметрии прядей. [35] (т.е. ограничение и но позволяя ).



Альтернативное обозначение также позволяет легко увидеть, как модель ОТО может быть применена к биологическим алфавитам с большим пространством состояний (например, аминокислотами или кодонами ). Набор частот состояний равновесия можно записать как , , ... и набор параметров взаимозаменяемости ( ) для любого алфавита состояния персонажа. Эти значения затем можно использовать для заполнения матрицу, установив недиагональные элементы, как показано выше (общее обозначение будет таким: ), установка диагональных элементов к отрицательной сумме недиагональных элементов в одной строке и нормализации. Очевидно, для аминокислот и для кодонов (при условии стандартного генетического кода ). Однако общность этих обозначений выгодна, поскольку для обозначения аминокислот можно использовать сокращенные алфавиты. Например, можно использовать и кодировать аминокислоты, перекодируя аминокислоты с использованием шести категорий, предложенных Маргарет Дэйхофф . Сокращенные аминокислотные алфавиты рассматриваются как способ уменьшить влияние композиционных вариаций и насыщенности. [36]









Важно отметить, что закономерности эволюции могут различаться в зависимости от региона генома, и, таким образом, разные регионы генома могут соответствовать разным моделям замещения. [37] Фактически, игнорирование гетерогенных закономерностей эволюции вдоль последовательностей может привести к систематическим ошибкам в оценке эволюционных параметров, включая K a /K s соотношение . В связи с этим использование смесевых моделей в филогенетических рамках удобно, чтобы лучше имитировать молекулярную эволюцию, наблюдаемую в реальных данных. [38]
Механистические и эмпирические модели
[ редактировать ]Основное различие между эволюционными моделями заключается в том, сколько параметров оценивается каждый раз для рассматриваемого набора данных и сколько из них оцениваются один раз для большого набора данных. Механистические модели описывают все замены как функцию ряда параметров, которые оцениваются для каждого анализируемого набора данных, предпочтительно с использованием максимального правдоподобия . Преимущество этого заключается в том, что модель можно адаптировать к особенностям конкретного набора данных (например, к различным отклонениям в составе ДНК). Проблемы могут возникнуть, когда используется слишком много параметров, особенно если они могут компенсировать друг друга (это может привести к неидентифицируемости). [39] ). Часто бывает так, что набор данных слишком мал, чтобы предоставить достаточно информации для точной оценки всех параметров.
Эмпирические модели создаются путем оценки многих параметров (обычно всех записей матрицы ставок, а также частот символов, см. модель GTR выше) из большого набора данных. Эти параметры затем фиксируются и будут повторно использоваться для каждого набора данных. Это имеет то преимущество, что эти параметры можно оценить более точно. Обычно невозможно оценить все элементы матрицы замены только на основе текущего набора данных. С другой стороны, параметры, оцененные на основе обучающих данных, могут быть слишком общими и, следовательно, плохо подходить для любого конкретного набора данных. Потенциальным решением этой проблемы является оценка некоторых параметров по данным с использованием метода максимального правдоподобия (или какого-либо другого метода). В исследованиях эволюции белков равновесные частоты аминокислот (с использованием однобуквенных кодов аминокислот IUPAC для обозначения их равновесных частот) часто оцениваются на основе данных [40] сохраняя при этом матрицу взаимозаменяемости фиксированной. Помимо общепринятой практики оценки частот аминокислот на основе данных, существуют методы оценки параметров обменности. [41] или отрегулируйте матрица [42] Для эволюции белков были предложены другие пути.

Поскольку крупномасштабное секвенирование генома по-прежнему дает очень большие объемы последовательностей ДНК и белков, имеется достаточно данных для создания эмпирических моделей с любым количеством параметров, включая эмпирические модели кодонов. [43] Из-за проблем, упомянутых выше, два подхода часто комбинируются путем однократной оценки большинства параметров на крупномасштабных данных, а несколько оставшихся параметров затем корректируются с учетом рассматриваемого набора данных. В следующих разделах представлен обзор различных подходов, используемых для моделей на основе ДНК, белков или кодонов.
Модели замены ДНК
[ редактировать ]Первые модели эволюции ДНК были предложены Джуксом и Кантором. [5] в 1969 году. Модель Джукса-Кантора (JC или JC69) предполагает равные скорости перехода, а также равные равновесные частоты для всех оснований, и это самая простая подмодель модели ОТО. В 1980 году компания Motoo Kimura представила модель с двумя параметрами (K2P или K80). [29] ): один для перехода и один для скорости трансверсии . Год спустя Кимура представил вторую модель (K3ST, K3P или K81). [31] ) с тремя типами замен: один для скорости перехода , другой для скорости трансверсий , сохраняющих сильные/слабые свойства нуклеотидов ( и , назначенный от Кимуры [31] ), и один для скорости трансверсий , которые сохраняют амино/кето-свойства нуклеотидов ( и , назначенный от Кимуры [31] ). В 1981 году Джозеф Фельзенштейн предложил четырехпараметрическую модель (F81 [28] ), в котором скорость замещения соответствует равновесной частоте целевого нуклеотида. Хасегава, Кишино и Яно объединили две последние модели в пятипараметрическую модель (HKY [30] ). После этих новаторских усилий в 1990-х годах в литературу (и широкое распространение) было введено множество дополнительных подмоделей модели GTR. [32] [33] Другие модели, которые определенным образом выходят за рамки модели ОТО, также были разработаны и усовершенствованы несколькими исследователями. [44] [45]




Почти все модели замещения ДНК являются механистическими моделями (как описано выше). Небольшое количество параметров, которые необходимо оценить для этих моделей, позволяет оценить эти параметры на основе данных. Это необходимо еще и потому, что закономерности эволюции последовательностей ДНК часто различаются между организмами и генами внутри организмов. Последнее может отражать оптимизацию путем отбора для конкретных целей (например, быстрая экспрессия или стабильность информационной РНК) или может отражать нейтральные вариации в паттернах замещения. Таким образом, в зависимости от организма и типа гена, вероятно, потребуется адаптировать модель к этим обстоятельствам.
Модели замещения с двумя состояниями
[ редактировать ]Альтернативный способ анализа данных о последовательностях ДНК — перекодировать нуклеотиды в пурины (R) и пиримидины (Y); [46] [47] эту практику часто называют RY-кодированием. [48] Вставки и удаления при множественном выравнивании последовательностей также могут быть закодированы как двоичные данные. [49] и проанализированы с использованием модели двух состояний. [50] [51]
Простейшая модель эволюции последовательности с двумя состояниями называется моделью Кавендера-Фарриса или моделью Кавендера-Фарриса- Неймана (CFN); Название этой модели отражает тот факт, что она была описана независимо в нескольких различных публикациях. [52] [53] [54] Модель CFN идентична модели Джукса-Кантора, адаптированной к двум состояниям, и она даже была реализована как модель «JC2» в популярном программном пакете IQ-TREE (использование этой модели в IQ-TREE требует кодирования данных как 0 и 1, а не R и Y; популярный пакет программного обеспечения PAUP* может интерпретировать матрицу данных, содержащую только R и Y, как данные для анализа с использованием модели CFN). Также легко анализировать двоичные данные с помощью филогенетического преобразования Адамара . [55] Альтернативная модель с двумя состояниями позволяет равновесным частотным параметрам R и Y (или 0 и 1) принимать значения, отличные от 0,5, путем добавления одного свободного параметра; эту модель по-разному называют CFu [46] или GTR2 (в IQ-TREE).
Модели замены аминокислот
[ редактировать ]Для многих анализов, особенно для более длинных эволюционных расстояний, эволюция моделируется на уровне аминокислот. Поскольку не все замены ДНК также изменяют кодируемую аминокислоту, информация теряется при просмотре аминокислот вместо нуклеотидных оснований. Однако несколько преимуществ говорят в пользу использования информации об аминокислотах: ДНК гораздо более склонна к смещению состава, чем аминокислоты, не все позиции в ДНК развиваются с одинаковой скоростью ( несинонимические мутации с меньшей вероятностью закрепляются в ДНК). популяция, чем синонимы ), но, вероятно, самое главное, из-за этих быстро развивающихся позиций и ограниченного размера алфавита (только четыре возможных состояния), ДНК страдает от большего количества обратных замен, что затрудняет точную оценку эволюционных расстояний.
В отличие от моделей ДНК, модели аминокислот традиционно являются эмпирическими моделями. Они были впервые применены в 1960-х и 1970-х годах Дейхоффом и его коллегами путем оценки скорости замещения на основе выравнивания белков с идентичностью не менее 85% (первоначально с очень ограниченными данными). [56] и, в конечном итоге, кульминацией которого стала модель Dayhoff PAM 1978 года. [57] ). Это минимизировало вероятность наблюдения множественных замен на сайте. Из оцененной матрицы скоростей была получена серия матриц вероятности замещения, известных под такими названиями, как PAM 250. Матрицы логарифмических шансов, основанные на модели Dayhoff PAM, обычно использовались для оценки значимости результатов поиска гомологии, хотя BLOSUM матрицы [58] в этом контексте заменили матрицы логарифмов шансов PAM , поскольку матрицы BLOSUM кажутся более чувствительными на различных эволюционных дистанциях, в отличие от PAM . матриц логарифмов шансов [59]
Матрица Дайхоффа PAM была источником параметров обменности, использованных в одном из первых анализов филогении с использованием метода максимального правдоподобия, в котором использовались данные о белках. [60] и модель PAM (или улучшенная версия модели PAM под названием DCMut). [61] ) продолжает использоваться в филогенетике. Однако ограниченное количество выравниваний, использованных для создания модели PAM (отражающее ограниченное количество данных о последовательностях, доступных в 1970-х годах), почти наверняка увеличило дисперсию некоторых параметров матрицы скоростей (альтернативно, белки, используемые для создания модели PAM, могли быть нерепрезентативный набор). Тем не менее, очевидно, что модель PAM редко так хорошо подходит для большинства наборов данных, как более современные эмпирические модели (Keane et al. 2006). [62] протестировали тысячи белков позвоночных , бактерий и архей и обнаружили, что модель Dayhoff PAM лучше всего подходит не более чем для <4% белков).
Начиная с 1990-х годов быстрое расширение баз данных последовательностей благодаря усовершенствованным технологиям секвенирования привело к оценке многих новых эмпирических матриц (см. [63] полный список). В самых ранних попытках использовались методы, аналогичные тем, которые использовал Дайхофф, с использованием крупномасштабного сопоставления базы данных белков для создания новой матрицы логарифмов шансов. [64] и модель JTT (Джонса-Тейлора-Торнтона). [65] Быстрый рост вычислительной мощности в это время (отражающий такие факторы, как закон Мура ) сделал возможным оценивать параметры эмпирических моделей с использованием метода максимального правдоподобия (например, WAG [40] и LG [66] модели) и другие методы (например, VT [67] и ПМБ [68] модели). Программный пакет IQ-Tree позволяет пользователям создавать собственные обратимые во времени модели с помощью QMaker. [69] или необратимое во времени с помощью nQMaker. [70]
Модель отсутствия общего механизма (NCM) и максимальная экономия
[ редактировать ]В 1997 году Таффли и Стил [71] описали модель, которую они назвали моделью без общего механизма (NCM). Топология дерева максимального правдоподобия для конкретного набора данных с учетом модели NCM идентична топологии оптимального дерева для тех же данных с учетом критерия максимальной экономии . Модель NCM предполагает, что все данные (например, гомологичные нуклеотиды, аминокислоты или морфологические признаки) связаны общим филогенетическим деревом. Затем параметры вводятся для каждого гомологичного признака, где это количество последовательностей. Это можно рассматривать как оценку отдельного параметра скорости для каждой пары символ × ветвь в наборе данных (обратите внимание, что количество ветвей в полностью разрешенном филогенетическом дереве равно ). Таким образом, количество свободных параметров в модели NCM всегда превышает количество гомологичных символов в матрице данных, а модель NCM подвергалась критике как постоянно «чрезмерно параметризованная». [72]


Ссылки
[ редактировать ]- ^ Jump up to: Перейти обратно: а б Стил М., Пенни Д. (июнь 2000 г.). «Экономность, правдоподобие и роль моделей в молекулярной филогенетике» . Молекулярная биология и эволюция . 17 (6): 839–850. doi : 10.1093/oxfordjournals.molbev.a026364 . ПМИД 10833190 .
- ^ Бромэм Л. (май 2019 г.). «Шесть невозможных вещей перед завтраком: предположения, модели и вера в молекулярное датирование». Тенденции в экологии и эволюции . 34 (5): 474–486. дои : 10.1016/j.tree.2019.01.017 . ПМИД 30904189 . S2CID 85496215 .
- ^ Ян З., Белявски Дж. П. (декабрь 2000 г.). «Статистические методы обнаружения молекулярной адаптации» . Тенденции в экологии и эволюции . 15 (12): 496–503. дои : 10.1016/s0169-5347(00)01994-7 . ПМЦ 7134603 . ПМИД 11114436 .
- ^ Перрон У., Козлов А.М., Стаматакис А., Гольдман Н., Моал И.Х. (сентябрь 2019 г.). Пупко Т (ред.). «Моделирование структурных ограничений эволюции белков через конформационные состояния боковой цепи» . Молекулярная биология и эволюция . 36 (9): 2086–2103. дои : 10.1093/molbev/msz122 . ПМК 6736381 . ПМИД 31114882 .
- ^ Jump up to: Перейти обратно: а б с д Джукс Т.Х., Кантор CH (1969). «Эволюция белковых молекул». В Манро HN (ред.). Белковый обмен млекопитающих . Том. 3. Эльзевир. стр. 21–132. дои : 10.1016/b978-1-4832-3211-9.50009-7 . ISBN 978-1-4832-3211-9 .
- ^ Хюльзенбек Дж. П., Хиллис Д. М. (1 сентября 1993 г.). «Успех филогенетических методов в случае четырех таксонов» . Систематическая биология . 42 (3): 247–264. дои : 10.1093/sysbio/42.3.247 . ISSN 1063-5157 .
- ^ Гольдман Н. (февраль 1993 г.). «Статистические испытания моделей замены ДНК». Журнал молекулярной эволюции . 36 (2): 182–198. Бибкод : 1993JMolE..36..182G . дои : 10.1007/BF00166252 . ПМИД 7679448 . S2CID 29354147 .
- ^ Суоффорд Д.Л. Олсен Г.Дж. Уодделл П.Дж. Хиллис Д.М. 1996. «Филогенетический вывод». в молекулярной систематике (под ред. Хиллиса Д.М. Морица К. Мейбл Б.К.) 2-е изд. Сандерленд, Массачусетс: Синауэр. п. 407–514. ISBN 978-0878932825
- ^ Черч С.Х., Райан Дж.Ф., Данн К.В. (ноябрь 2015 г.). «Автоматизация и оценка теста SOWH с помощью SOWHAT» . Систематическая биология . 64 (6): 1048–1058. дои : 10.1093/sysbio/syv055 . ПМК 4604836 . ПМИД 26231182 .
- ^ Льюис ПО (01 ноября 2001 г.). «Правдоподобный подход к оценке филогении на основе дискретных данных о морфологических признаках» . Систематическая биология . 50 (6): 913–925. дои : 10.1080/106351501753462876 . ПМИД 12116640 .
- ^ Ли М.С., Кау А., Нэйш Д., Дайк Г.Дж. (май 2014 г.). «Морфологические часы в палеонтологии и среднемеловое происхождение короны Авеса» . Систематическая биология . 63 (3): 442–449. дои : 10.1093/sysbio/syt110 . ПМИД 24449041 .
- ^ Ронквист Ф., Клопфштейн С., Вильгельмсен Л., Шульмейстер С., Мюррей Д.Л., Расницын А.П. (декабрь 2012 г.). «Подход с полным доказательством датирования окаменелостей применительно к ранней радиации перепончатокрылых» . Систематическая биология . 61 (6): 973–999. дои : 10.1093/sysbio/sys058 . ПМЦ 3478566 . ПМИД 22723471 .
- ^ Брауэр, AV.Z. (2016). «Мы все кладисты?» у Уильямса Д., Шмитта М. и Уиллера К. (ред.). Будущее филогенетической систематики: наследие Вилли Хеннига (Книга 86 специальной серии Ассоциации систематики). Издательство Кембриджского университета. стр. 88-114 ISBN 978-1107117648
- ^ Фаррис Дж.С., Клюге А.Г., Карпентер Дж.М. (июнь 2001 г.). Олмстед Р. (ред.). «Поппер и вероятность против «Поппера» » . Систематическая биология . 50 (3): 438–444. дои : 10.1080/10635150119150 . ПМИД 12116585 .
- ^ Голдман, Ник (декабрь 1990 г.). «Вывод о максимальном правдоподобии филогенетических деревьев с особым упором на модель пуассоновского процесса замены ДНК и экономный анализ» . Систематическая зоология . 39 (4): 345–361. дои : 10.2307/2992355 . JSTOR 2992355 .
- ^ Гу X, Ли WH (сентябрь 1992 г.). «Более высокие показатели замены аминокислот у грызунов, чем у людей» . Молекулярная филогенетика и эволюция . 1 (3): 211–214. дои : 10.1016/1055-7903(92)90017-Б . ПМИД 1342937 .
- ^ Ли В.Х., Эллсуорт Д.Л., Крушкал Дж., Чанг Б.Х., Хьюитт-Эмметт Д. (февраль 1996 г.). «Скорость замены нуклеотидов у приматов и грызунов и гипотеза эффекта времени генерации». Молекулярная филогенетика и эволюция . 5 (1): 182–187. дои : 10.1006/mpev.1996.0012 . ПМИД 8673286 .
- ^ Мартин А.П., Палумби С.Р. (май 1993 г.). «Размер тела, скорость метаболизма, время генерации и молекулярные часы» . Труды Национальной академии наук Соединенных Штатов Америки . 90 (9): 4087–4091. Бибкод : 1993PNAS...90.4087M . дои : 10.1073/pnas.90.9.4087 . ПМК 46451 . ПМИД 8483925 .
- ^ Ян З., Нильсен Р. (апрель 1998 г.). «Синонимичные и несинонимичные изменения скорости ядерных генов млекопитающих». Журнал молекулярной эволюции . 46 (4): 409–418. Бибкод : 1998JMolE..46..409Y . CiteSeerX 10.1.1.19.7744 . дои : 10.1007/PL00006320 . ПМИД 9541535 . S2CID 13917969 .
- ^ Кишино Х., Торн Дж.Л., Бруно В.Дж. (март 2001 г.). «Эффективность метода оценки времени дивергенции в рамках вероятностной модели эволюции скорости» . Молекулярная биология и эволюция . 18 (3): 352–361. doi : 10.1093/oxfordjournals.molbev.a003811 . ПМИД 11230536 .
- ^ Торн Дж.Л., Кишино Х., Художник И.С. (декабрь 1998 г.). «Оценка скорости эволюции скорости молекулярной эволюции» . Молекулярная биология и эволюция . 15 (12): 1647–1657. doi : 10.1093/oxfordjournals.molbev.a025892 . ПМИД 9866200 .
- ^ Jump up to: Перейти обратно: а б с Таваре С. «Некоторые вероятностные и статистические проблемы анализа последовательностей ДНК» (PDF) . Лекции по математике в науках о жизни . 17 : 57–86.
- ^ Jump up to: Перейти обратно: а б Ян Цзы (2006). Вычислительная молекулярная эволюция . Оксфорд: Издательство Оксфордского университета. ISBN 978-1-4294-5951-8 . OCLC 99664975 .
- ^ Jump up to: Перейти обратно: а б с Ян Цзы (июль 1994 г.). «Оценка характера замены нуклеотидов». Журнал молекулярной эволюции . 39 (1): 105–111. Бибкод : 1994JMolE..39..105Y . дои : 10.1007/BF00178256 . PMID 8064867 . S2CID 15895455 .
- ^ Суоффорд, Д.Л., Олсен, Г.Дж., Уодделл, П.Дж. и Хиллис, Д.М. (1996) Филогенетический вывод. В: Хиллис, Д.М., Мориц, К. и Мейбл, Б.К., ред., Молекулярная систематика, 2-е издание, Sinauer Associates, Сандерленд (Массачусетс), 407-514. ISBN 0878932828 ISBN 978-0878932825
- ^ Фельзенштейн Дж (2004). Выводы о филогениях . Сандерленд, Массачусетс: Sinauer Associates. ISBN 0-87893-177-5 . OCLC 52127769 .
- ^ Суоффорд Д.Л., Bell CD (1997). «(Проект) руководства ПАУП*» . Проверено 31 декабря 2019 г.
- ^ Jump up to: Перейти обратно: а б с Фельзенштейн Дж. (ноябрь 1981 г.). «Эволюционные деревья на основе последовательностей ДНК: подход максимального правдоподобия». Журнал молекулярной эволюции . 17 (6): 368–376. Бибкод : 1981JMolE..17..368F . дои : 10.1007/BF01734359 . ПМИД 7288891 . S2CID 8024924 .
- ^ Jump up to: Перейти обратно: а б Кимура М. (декабрь 1980 г.). «Простой метод оценки скорости эволюции оснований посредством сравнительных исследований нуклеотидных последовательностей». Журнал молекулярной эволюции . 16 (2): 111–120. Бибкод : 1980JMolE..16..111K . дои : 10.1007/BF01731581 . ПМИД 7463489 . S2CID 19528200 .
- ^ Jump up to: Перейти обратно: а б Хасэгава М., Кишино Х., Яно Т. (октябрь 1985 г.). «Датирование расщепления человека и обезьяны по молекулярным часам митохондриальной ДНК». Журнал молекулярной эволюции . 22 (2): 160–174. Бибкод : 1985JMolE..22..160H . дои : 10.1007/BF02101694 . ПМИД 3934395 . S2CID 25554168 .
- ^ Jump up to: Перейти обратно: а б с д Кимура М. (январь 1981 г.). «Оценка эволюционных расстояний между гомологичными нуклеотидными последовательностями» . Труды Национальной академии наук Соединенных Штатов Америки . 78 (1): 454–458. Бибкод : 1981PNAS...78..454K . дои : 10.1073/pnas.78.1.454 . ПМК 319072 . ПМИД 6165991 .
- ^ Jump up to: Перейти обратно: а б Тамура К., Ней М. (май 1993 г.). «Оценка количества нуклеотидных замен в контрольной области митохондриальной ДНК человека и шимпанзе» . Молекулярная биология и эволюция . 10 (3): 512–526. doi : 10.1093/oxfordjournals.molbev.a040023 . ПМИД 8336541 .
- ^ Jump up to: Перейти обратно: а б с Жарких А. (сентябрь 1994 г.). «Оценка эволюционных расстояний между нуклеотидными последовательностями». Журнал молекулярной эволюции . 39 (3): 315–329. Бибкод : 1994JMolE..39..315Z . дои : 10.1007/BF00160155 . ПМИД 7932793 . S2CID 33845318 .
- ^ Хюльзенбек Дж. П., Ларжет Б., Альфаро М. Е. (июнь 2004 г.). «Выбор байесовской филогенетической модели с использованием обратимой скачкообразной цепи Маркова Монте-Карло» . Молекулярная биология и эволюция . 21 (6): 1123–1133. дои : 10.1093/molbev/msh123 . ПМИД 15034130 .
- ^ Яп В.Б., Пахтер Л. (апрель 2004 г.). «Идентификация эволюционных горячих точек в геномах грызунов» . Геномные исследования . 14 (4): 574–579. дои : 10.1101/гр.1967904 . ПМЦ 383301 . ПМИД 15059998 .
- ^ Суско Э., Роджер А.Дж. (сентябрь 2007 г.). «О сокращенных аминокислотных алфавитах для филогенетических выводов» . Молекулярная биология и эволюция . 24 (9): 2139–2150. дои : 10.1093/molbev/msm144 . ПМИД 17652333 .
- ^ Арбиса, Леонардо; Патрисио, Матеус; Допазо, Эрнан; Посада, Дэвид (1 января 2011 г.). «Общегеномная гетерогенность подходящей модели нуклеотидного замещения» . Геномная биология и эволюция . 3 : 896–908. дои : 10.1093/gbe/evr080 . ISSN 1759-6653 . ПМК 3175760 . ПМИД 21824869 .
- ^ Халперн, Алабама; Бруно, WJ (1 июля 1998 г.). «Эволюционные расстояния для последовательностей, кодирующих белок: моделирование частот сайт-специфических остатков» . Молекулярная биология и эволюция . 15 (7): 910–917. doi : 10.1093/oxfordjournals.molbev.a025995 . ISSN 0737-4038 . ПМИД 9656490 .
- ^ Пончано Дж. М., Берли Дж. Г., Браун Э. Л., Тапер М. Л. (декабрь 2012 г.). «Оценка идентифицируемости параметров в филогенетических моделях с использованием клонирования данных» . Систематическая биология . 61 (6): 955–972. дои : 10.1093/sysbio/sys055 . ПМЦ 3478565 . ПМИД 22649181 .
- ^ Jump up to: Перейти обратно: а б Уилан С., Голдман Н. (май 2001 г.). «Общая эмпирическая модель эволюции белков, полученная на основе нескольких семейств белков с использованием подхода максимального правдоподобия» . Молекулярная биология и эволюция . 18 (5): 691–699. doi : 10.1093/oxfordjournals.molbev.a003851 . ПМИД 11319253 .
- ^ Браун Э.Л. (июль 2018 г.). «Эволюционная модель, основанная на физико-химических свойствах аминокислот, обнаруживает различия между белками» . Биоинформатика . 34 (13): i350–i356. doi : 10.1093/биоинформатика/bty261 . ПМК 6022633 . ПМИД 29950007 .
- ^ Голдман Н., Уилан С. (ноябрь 2002 г.). «Новое использование равновесных частот в моделях эволюции последовательностей» . Молекулярная биология и эволюция . 19 (11): 1821–1831. doi : 10.1093/oxfordjournals.molbev.a004007 . ПМИД 12411592 .
- ^ Косиол С., Холмс И., Голдман Н. (июль 2007 г.). «Эмпирическая модель кодонов для эволюции белковой последовательности» . Молекулярная биология и эволюция . 24 (7): 1464–1479. дои : 10.1093/molbev/msm064 . ПМИД 17400572 .
- ^ Тамура К. (июль 1992 г.). «Оценка количества нуклеотидных замен при наличии сильных смещений переход-трансверсия и содержание G + C» . Молекулярная биология и эволюция . 9 (4): 678–687. doi : 10.1093/oxfordjournals.molbev.a040752 . ПМИД 1630306 .
- ^ Халперн А.Л., Бруно В.Дж. (июль 1998 г.). «Эволюционные расстояния для последовательностей, кодирующих белок: моделирование частот сайт-специфических остатков» . Молекулярная биология и эволюция . 15 (7): 910–917. doi : 10.1093/oxfordjournals.molbev.a025995 . ПМИД 9656490 . S2CID 7332698 .
- ^ Jump up to: Перейти обратно: а б Браун Э.Л., Кимбалл Р.Т. (август 2002 г.). Кьер К. (ред.). «Изучение базальных птичьих расхождений с помощью митохондриальных последовательностей: сложность модели, выборка таксонов и длина последовательности» . Систематическая биология . 51 (4): 614–625. дои : 10.1080/10635150290102294 . ПМИД 12228003 .
- ^ Филлипс М.Дж., Дельсук Ф., Пенни Д. (июль 2004 г.). «Филогения в масштабе генома и обнаружение систематических ошибок» . Молекулярная биология и эволюция . 21 (7): 1455–1458. дои : 10.1093/molbev/msh137 . ПМИД 15084674 .
- ^ Исикава С.А., Инагаки Ю, Хашимото Т (январь 2012 г.). «RY-кодирование и негомогенные модели могут улучшить выводы максимального правдоподобия на основе данных нуклеотидных последовательностей с параллельной композиционной неоднородностью» . Эволюционная биоинформатика онлайн . 8 : 357–371. дои : 10.4137/EBO.S9017 . ПМЦ 3394461 . ПМИД 22798721 .
- ^ Член парламента Симмонса, Охотерена Х (июнь 2000 г.). «Пробелы как символы в филогенетическом анализе на основе последовательностей» . Систематическая биология . 49 (2): 369–381. дои : 10.1093/sysbio/49.2.369 . ПМИД 12118412 .
- ^ Юрий Т., Кимбалл Р.Т., Харшман Дж., Боуи Р.К., Браун М.Дж., Хойновски Дж.Л. и др. (март 2013 г.). «Экономный и основанный на моделях анализ инделей в ядерных генах птиц выявляет конгруэнтные и неконгруэнтные филогенетические сигналы» . Биология . 2 (1): 419–444. дои : 10.3390/biology2010419 . ПМК 4009869 . ПМИД 24832669 .
- ^ Уд П., Браун Э.Л., Нарула Н., Минхарес У., Мирараб С. (06.07.2019). «Филогенетический сигнал инделов и неавианской радиации» . Разнообразие . 11 (7): 108. дои : 10.3390/d11070108 .
- ^ Кавендер Дж. А. (август 1978 г.). «Таксономия с уверенностью». Математические биологические науки . 40 (3–4): 271–280. дои : 10.1016/0025-5564(78)90089-5 .
- ^ Фаррис Дж.С. (1 сентября 1973 г.). «Вероятностная модель для построения эволюционных деревьев» . Систематическая биология . 22 (3): 250–256. дои : 10.1093/sysbio/22.3.250 . ISSN 1063-5157 .
- ^ Нейман Дж (1971). Гупта СС, Якель Дж (ред.). Молекулярные исследования эволюции: источник новых статистических проблем . Нью-Йорк, штат Нью-Йорк, США: New York Academic Press. стр. 1–27.
- ^ Уодделл П.Дж., Пенни Д., Мур Т. (август 1997 г.). «Сопряжения Адамара и моделирование эволюции последовательностей с неравными скоростями на разных сайтах». Молекулярная филогенетика и эволюция . 8 (1): 33–50. дои : 10.1006/mpev.1997.0405 . ПМИД 9242594 .
- ^ Дайхофф М.О., Эк Р.В., Парк К.М. (1969). «Модель эволюционного изменения белков» . В Дайхоффе, Миссури (ред.). Атлас последовательности и структуры белка . Том. 4. С. 75–84.
- ^ Дайхофф М.О., Шварц Р.М., Оркатт, Британская Колумбия (1978). «Модель эволюционных изменений белков» (PDF) . В Дайхоффе, Миссури (ред.). Атлас последовательности и структуры белка . Том. 5. С. 345–352.
- ^ Хеникофф С., Хеникофф Дж.Г. (ноябрь 1992 г.). «Матрицы аминокислотных замен из белковых блоков» . Труды Национальной академии наук Соединенных Штатов Америки . 89 (22): 10915–10919. Бибкод : 1992PNAS...8910915H . дои : 10.1073/pnas.89.22.10915 . ПМК 50453 . ПМИД 1438297 .
- ^ Альтшул С.Ф. (март 1993 г.). «Система оценки выравнивания белков, чувствительная на всех эволюционных расстояниях». Журнал молекулярной эволюции . 36 (3): 290–300. Бибкод : 1993JMolE..36..290A . дои : 10.1007/BF00160485 . ПМИД 8483166 . S2CID 22532856 .
- ^ Кишино Х., Мията Т., Хасэгава М. (август 1990 г.). «Вывод максимального правдоподобия о филогении белков и происхождении хлоропластов». Журнал молекулярной эволюции . 31 (2): 151–160. Бибкод : 1990JMolE..31..151K . дои : 10.1007/BF02109483 . S2CID 24650412 .
- ^ Косиол С., Гольдман Н. (февраль 2005 г.). «Различные версии матрицы ставок Дайхоффа» . Молекулярная биология и эволюция . 22 (2): 193–199. дои : 10.1093/molbev/msi005 . ПМИД 15483331 .
- ^ Кин Т.М., Криви С.Дж., Пентони М.М., Нотон Т.Дж., Маклнерни Дж.О. (март 2006 г.). «Оценка методов выбора аминокислотной матрицы и их использования на эмпирических данных показывает, что специальные предположения по выбору матрицы не оправданы» . Эволюционная биология BMC . 6 (1): 29. Бибкод : 2006BMCEE...6...29K . дои : 10.1186/1471-2148-6-29 . ПМЦ 1435933 . ПМИД 16563161 .
- ^ Биго Т., Гульельмини Дж., Крискуоло А. (август 2019 г.). «Данные моделирования для оценки числовых констант для аппроксимации попарных эволюционных расстояний между аминокислотными последовательностями» . Данные вкратце . 25 : 104212. Цифровой код : 2019DIB....2504212B . дои : 10.1016/j.dib.2019.104212 . ПМК 6699465 . ПМИД 31440543 .
- ^ Гонне Г.Х., Коэн М.А., Беннер С.А. (июнь 1992 г.). «Исчерпывающее сопоставление всей базы данных последовательностей белков». Наука . 256 (5062): 1443–1445. Бибкод : 1992Sci...256.1443G . дои : 10.1126/science.1604319 . ПМИД 1604319 .
- ^ Джонс Д.Т., Тейлор В.Р., Торнтон Дж.М. (июнь 1992 г.). «Быстрое создание матриц данных о мутациях из белковых последовательностей». Компьютерные приложения в биологических науках . 8 (3): 275–282. дои : 10.1093/биоинформатика/8.3.275 . ПМИД 1633570 .
- ^ Le SQ, Гаскюэль О (июль 2008 г.). «Улучшенная матрица замены общих аминокислот» . Молекулярная биология и эволюция . 25 (7): 1307–1320. дои : 10.1093/molbev/msn067 . ПМИД 18367465 .
- ^ Мюллер Т., Вингрон М. (декабрь 2000 г.). «Моделирование аминокислотной замены». Журнал вычислительной биологии . 7 (6): 761–776. дои : 10.1089/10665270050514918 . ПМИД 11382360 .
- ^ Вирассами С., Смит А., Тилье Э.Р. (декабрь 2003 г.). «Модель вероятности перехода для аминокислотных замен из блоков». Журнал вычислительной биологии . 10 (6): 997–1010. дои : 10.1089/106652703322756195 . ПМИД 14980022 .
- ^ Минь, Буй Куанг; Данг, Куонг Цао; Винь, Ле Си; Ланфир, Роберт (11 августа 2021 г.). «QMaker: быстрый и точный метод оценки эмпирических моделей эволюции белков» . Систематическая биология . 70 (5): 1046–1060. дои : 10.1093/sysbio/syab010 . ПМЦ 8357343 . ПМИД 33616668 .
- ^ Данг, Куонг Цао; Минь, Буй Куанг; Макши, Ханон; Мазель, Джоанна; Джеймс, Дженнифер Элеонора; Винь, Ле Си; Ланфир, Роберт (9 февраля 2022 г.). «nQMaker: оценка времени моделей необратимой замены аминокислот» . Систематическая биология . 71 (5): 1110–1123. дои : 10.1093/sysbio/syac007 . ПМЦ 9366462 . ПМИД 35139203 .
- ^ Таффли С., Стил М. (май 1997 г.). «Связь между максимальной вероятностью и максимальной экономией в рамках простой модели замены сайтов». Бюллетень математической биологии . 59 (3): 581–607. дои : 10.1007/bf02459467 . ПМИД 9172826 . S2CID 189885872 .
- ^ Холдер М.Т., Льюис П.О., Суоффорд Д.Л. (июль 2010 г.). «Информационный критерий akaike не выберет модель отсутствия общего механизма» . Систематическая биология . 59 (4): 477–485. дои : 10.1093/sysbio/syq028 . hdl : 1808/9209 . ПМИД 20547783 .
Хорошая модель для филогенетического вывода должна быть достаточно богатой, чтобы справиться с источниками шума в данных, но оценка ML, проводимая с использованием моделей, которые явно чрезмерно параметризованы, может привести к совершенно неверным выводам. Модель NCM, безусловно, попадает в область слишком большого количества параметров, чтобы служить оправданием использования экономии, основанной на том, что она является оценщиком ML в рамках общей модели.
Внешние ссылки
[ редактировать ]Примечания
[ редактировать ]- ^ Ссылка описывает полемику #ParsimonyGate, которая представляет собой конкретный пример дебатов относительно философской природы критерия максимальной экономности. #ParsimonyGate стал реакцией в Твиттере на редакционную статью в журнале Cladistics, опубликованном Обществом Вилли Хеннига. В редакционной статье говорится, что «... эпистемологическая парадигма этого журнала - это экономия», и утверждается, что существуют философские причины предпочитать экономность другим методам филогенетического вывода. Поскольку другие методы филогенетического вывода (например, метод максимального правдоподобия, байесовский вывод, филогенетические инварианты и большинство дистанционных методов) основаны на моделях, это утверждение неявно отвергает представление о том, что экономия является моделью.