Формат FASTQ
| Тип интернет-СМИ | текстовый/обычный |
|---|---|
| Разработано | Wellcome Trust Sanger Institute |
| Первоначальный выпуск | ~2000 |
| Тип формата | Биоинформатика |
| Расширено с | ASCII и FASTA. Формат |
| Веб-сайт | журнал |
Формат FASTQ — это текстовый формат для хранения как биологической последовательности (обычно нуклеотидной последовательности ), так и соответствующих ей показателей качества. И буква последовательности, и показатель качества для краткости кодируются одним символом ASCII .
Первоначально он был разработан в Wellcome Trust Sanger Institute для объединения последовательности в формате FASTA и данных о ее качестве, но стал фактическим стандартом для хранения результатов высокопроизводительных инструментов секвенирования, таких как анализатор генома Illumina . [ 1 ]
Формат
[ редактировать ]Файл FASTQ имеет четыре поля, разделенных строками на последовательность:
- Поле 1 начинается с символа «@», за которым следует идентификатор последовательности и необязательное описание (например, строка заголовка FASTA ).
- Поле 2 — это необработанные буквы последовательности.
- Поле 3 начинается с символа «+», за которым при необходимости следует еще раз тот же идентификатор последовательности (и любое описание).
- Поле 4 кодирует значения качества для последовательности в Поле 2 и должно содержать то же количество символов, что и буквы в последовательности.
Файл FASTQ, содержащий одну последовательность, может выглядеть так:
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Байт, обозначающий качество, варьируется от 0x21 (самое низкое качество; «!» в ASCII) до 0x7e (самое высокое качество; «~» в ASCII). Вот символы значения качества в порядке возрастания качества слева направо ( ASCII ):
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
Исходные файлы Sanger FASTQ разбивают длинные последовательности и строки качества на несколько строк, как это обычно делается для FASTA файлов . Учет этого усложняет синтаксический анализ из-за выбора «@» и «+» в качестве маркеров (поскольку эти символы также могут встречаться в строке качества). Многострочные файлы FASTQ (и, следовательно, многострочные анализаторы FASTQ) стали менее распространены в настоящее время, поскольку большая часть выполняемого секвенирования представляет собой секвенирование Illumina с коротким чтением с типичной длиной последовательности около 100 пар оснований.
Идентификаторы последовательностей Illumina
[ редактировать ]Последовательности из программного обеспечения Illumina используют систематический идентификатор:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | уникальное название инструмента |
|---|---|
| 6 | полоса проточной ячейки |
| 73 | номер плитки на полосе проточной кюветы |
| 941 | 'x' — координата кластера внутри тайла |
| 1973 | 'y' — координата кластера внутри тайла |
| #0 | порядковый номер для мультиплексированной выборки (0 — без индексации) |
| /1 | член пары /1 или /2 (парный конец или сопряженная пара только для чтения) |
Версии конвейера Illumina, начиная с версии 1.4, используют #NNNNNN вместо #0 для идентификатора мультиплекса, где NNNNNN — это последовательность тега мультиплекса.
В Casava 1.8 формат строки @ изменился:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | уникальное название инструмента |
|---|---|
| 136 | идентификатор запуска |
| FC706VJ | идентификатор проточной ячейки |
| 2 | полоса проточной ячейки |
| 2104 | номер плитки на полосе проточной кюветы |
| 15343 | 'x' — координата кластера внутри тайла |
| 197393 | 'y' — координата кластера внутри тайла |
| 1 | член пары, 1 или 2 (парный конец или сопряженная пара только читает) |
| И | Y, если чтение отфильтровано (не прошло), N в противном случае |
| 18 | 0, когда ни один из битов управления не включен, в противном случае это четное число |
| АТСАКГ | индексная последовательность |
Обратите внимание, что более поздние версии программного обеспечения Illumina выводят номер образца (определяемый порядком образцов в протоколе проб) вместо индексной последовательности, если индексная последовательность явно не указана для образца в протоколе проб. Например, следующий заголовок может появиться в файле FASTQ, принадлежащем первому образцу из пакета образцов:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:N:18:1
Архив чтения последовательностей NCBI
[ редактировать ]Файлы FASTQ из INSDC архива чтения последовательностей часто содержат описание, например
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
В этом примере имеется идентификатор, назначенный NCBI, а описание содержит исходный идентификатор от Solexa/Illumina (как описано выше) плюс длину считывания. Секвенирование проводили в парном режиме (размер вставки ~500 п.н.), см. SRR001666 . Формат вывода fastq-dump по умолчанию создает целые фрагменты, содержащие любые технические чтения и, как правило, биологические чтения с одиночным или парным концом.
$ fastq-dump.2.9.0 -Z -X 2 SRR001666
Read 2 spots for SRR001666
Written 2 spots for SRR001666
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
Современное использование FASTQ почти всегда предполагает разделение пятна на его биологические чтения, как описано в метаданных, предоставленных отправителем:
$ fastq-dump -X 2 SRR001666 --split-3
Read 2 spots for SRR001666
Written 2 spots for SRR001666
$ head SRR001666_1.fastq SRR001666_2.fastq
==> SRR001666_1.fastq <==
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
==> SRR001666_2.fastq <==
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36
IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
Если fastq-dump присутствует в архиве, он может попытаться восстановить прочитанные имена в исходный формат. По умолчанию NCBI не сохраняет исходные прочитанные имена:
$ fastq-dump -X 2 SRR001666 --split-3 --origfmt
Read 2 spots for SRR001666
Written 2 spots for SRR001666
$ head SRR001666_1.fastq SRR001666_2.fastq
==> SRR001666_1.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@071112_SLXA-EAS1_s_7:5:1:801:338
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI
==> SRR001666_2.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@071112_SLXA-EAS1_s_7:5:1:801:338
AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
В приведенном выше примере использовались исходные имена чтения, а не присоединенное имя чтения. Запуски NCBI-доступа и содержащиеся в них чтения. Исходные имена чтения, присвоенные секвенсорами, могут функционировать как локальные уникальные идентификаторы чтения и передавать ровно столько же информации, сколько и серийный номер. Приведенные выше идентификаторы были назначены алгоритмически на основе информации о пробеге и геометрических координатах. Ранние загрузчики SRA анализировали эти идентификаторы и сохраняли их разложенные компоненты внутри себя. NCBI прекратил записывать имена чтения, поскольку они часто изменяются по сравнению с исходным форматом поставщиков, чтобы связать некоторую дополнительную информацию, имеющую значение для конкретного конвейера обработки, и это вызвало нарушения формата имени, которые привели к большому количеству отклоненных заявок. Без четкой схемы имен чтения их функция остается уникальной идентификатором чтения, передающей тот же объем информации, что и прочитанный серийный номер. см. в различных выпусках SRA Toolkit Подробности и обсуждения .
Также обратите внимание, что fastq-dump преобразует эти данные FASTQ из исходной кодировки Solexa/Illumina в стандарт Sanger (см. кодировки ниже). Это связано с тем, что SRA служит хранилищем информации NGS, а не формата . Различные инструменты *-dump способны создавать данные в нескольких форматах из одного источника. Требования для этого диктовались пользователями на протяжении нескольких лет, при этом большая часть раннего спроса исходила от проекта «1000 геномов» .
Вариации
[ редактировать ]Качество
[ редактировать ]Значение качества Q представляет собой целочисленное отображение p (т. е. вероятность того, что соответствующий базовый вызов неверен). Использовались два разных уравнения. Первый — это стандартный вариант Сэнгера для оценки надежности базового вызова, также известный как показатель качества Phred :

Конвейер Solexa (т. е. программное обеспечение, поставляемое с анализатором генома Illumina) ранее использовал другое сопоставление, кодируя шансы p /(1- p ) вместо вероятности p :

Хотя оба отображения асимптотически идентичны при более высоких значениях качества, они различаются при более низких уровнях качества (т. е. примерно p > 0,05 или, что эквивалентно, Q < 13).
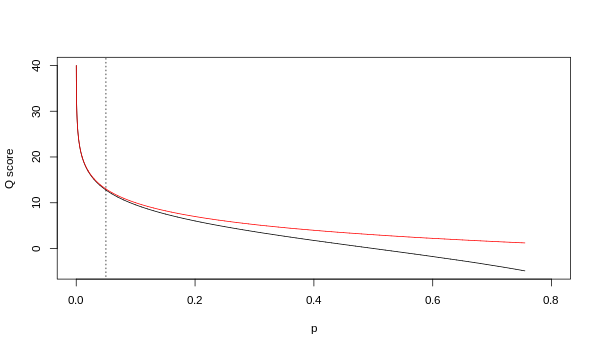
Время от времени возникали разногласия по поводу того, какие карты на самом деле использует Illumina. В руководстве пользователя (Приложение B, стр. 122) для версии 1.4 конвейера Illumina указано, что: «Оценки определяются как [ sic ], где p — вероятность вызова базы, соответствующей рассматриваемой базе». [ 2 ] Оглядываясь назад, можно сказать, что эта запись в руководстве была ошибкой. В руководстве пользователя («Что нового», стр. 5) для версии 1.5 конвейера Illumina вместо этого указано следующее описание: «Важные изменения в конвейере v1.3 [ sic ]. Схема оценки качества изменилась на схему оценки Phred [т. е. Сэнгера] , закодированный как символ ASCII путем добавления 64 к значению Phred. Оценка Phred базы равна: , где e — предполагаемая вероятность неправильного основания. [ 3 ]


Кодирование
[ редактировать ]- Формат Сэнгера может кодировать оценку качества Phred от 0 до 93 с использованием ASCII от 33 до 126 (хотя в необработанных данных чтения показатель качества Phred редко превышает 60, более высокие оценки возможны в сборках или картах чтения). Также используется в формате SAM. [ 4 ] Согласно объявлению на форуме seqanswers.com, к концу февраля 2011 года новейшая версия конвейера CASAVA компании Illumina (1.8) будет напрямую создавать fastq в формате Sanger. [ 5 ]
- Чтения AVITI компании Element Biosciences кодируются в соответствии с соглашением Сэнгера: оценки качества Phred от 0 до 93 кодируются с использованием ASCII от 33 до 126. Необработанные чтения обычно имеют базовые оценки качества в диапазоне [0, 55]. [ 6 ]
- Чтения PacBio HiFi, которые обычно хранятся в формате SAM/BAM, используют соглашение Сэнгера: оценки качества Phred от 0 до 93 кодируются с использованием ASCII от 33 до 126. Необработанные подчтения PacBio используют то же соглашение, но обычно назначают заполнитель базового качества (Q0). ) ко всем базам в прочтении. [ 7 ]
- Чтения Oxford Nanopore Duplex, вызываемые с использованием базового вызывающего устройства Dorado, обычно сохраняются в формате SAM/BAM. После перехода на 16-битное внутреннее представление качества заявленный базовый предел качества составляет q50 (S). [ 8 ]
- Формат Solexa/Illumina 1.0 может кодировать показатель качества Solexa/Illumina от -5 до 62 с использованием ASCII от 59 до 126 (хотя в необработанных считанных данных ожидаются только оценки Solexa от -5 до 40).
- Начиная с Illumina 1.3 и до Illumina 1.8, в этом формате показатель качества Phred кодировался от 0 до 62 с использованием ASCII от 64 до 126 (хотя в необработанных считанных данных ожидаются только оценки Phred от 0 до 40).
- Начиная с Illumina 1.5 и до Illumina 1.8, значения Phred от 0 до 2 имеют немного другое значение. Значения 0 и 1 больше не используются, а значение 2, закодированное ASCII 66 «B», также используется в конце чтения в качестве индикатора контроля качества сегмента чтения . [ 9 ] Руководство Иллюмины [ 10 ] (стр. 30) гласит следующее: Если чтение заканчивается сегментом преимущественно низкого качества (Q15 или ниже), то все значения качества в сегменте заменяются значением 2 (закодировано буквой B в тексте Illumina). -кодирование показателей качества)... Этот показатель Q2 не прогнозирует конкретную частоту ошибок, а скорее указывает на то, что конкретная заключительная часть считывания не должна использоваться в дальнейшем анализе. Кроме того, показатель качества, закодированный буквой «B», может встречаться внутри операций чтения, по крайней мере, начиная с версии конвейера 1.6, как показано в следующем примере:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT +HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1 efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
Была предложена альтернативная интерпретация этой кодировки ASCII. [ 11 ] Кроме того, при прогоне Illumina с использованием элементов управления PhiX было замечено, что символ «B» представляет «неизвестный показатель качества». Частота ошибок чтения «B» была примерно на 3 балла ниже среднего наблюдаемого балла для данного запуска.
- Начиная с Illumina 1.8, показатели качества в основном вернулись к использованию формата Сэнгера (Phred+33).
Для необработанных считываний диапазон оценок будет зависеть от используемой технологии и вызывающего основания, но обычно составляет до 41 для последних химических анализов Illumina. Поскольку максимальный наблюдаемый показатель качества ранее составлял всего 40, различные сценарии и инструменты ломаются, когда сталкиваются с данными со значениями качества, превышающими 40. Для обработанных операций чтения оценки могут быть еще выше. Например, значения качества 45 наблюдаются при чтении службы секвенирования длинного чтения Illumina (ранее Moleculo).
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX...................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... .................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ..................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN........................................... EEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEE PPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPP !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | | 33 59 64 73 88 104 126 0........................26...31.......40 -5....0........9.............................40 0........9.............................40 3.....9..............................41 0.2......................26...31........41 0..................20........30........40........50 0..................20........30........40........50...55 0..................20........30........40........50..........................................93
S - Sanger Phred+33, raw reads typically (0, 40) X - Solexa Solexa+64, raw reads typically (-5, 40) I - Illumina 1.3+ Phred+64, raw reads typically (0, 40) J - Illumina 1.5+ Phred+64, raw reads typically (3, 41) with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold) (Note: See discussion above). L - Illumina 1.8+ Phred+33, raw reads typically (0, 41) N - Nanopore Phred+33, Duplex reads typically (0, 50) E - ElemBio AVITI Phred+33, raw reads typically (0, 55) P - PacBio Phred+33, HiFi reads typically (0, 93)
Цветовое пространство
[ редактировать ]Для данных SOLiD формат изменяется на последовательность FASTQ цветового пространства (CSFASTQ), где основания в последовательности комбинируются с числами 0, 1, 2 и 3, указывая, как основания изменяются относительно предыдущего основания в последовательности. (0: без изменений; 1: переход; 2: некомплементарная трансверсия; 3: комплементарная трансверсия). [ 1 ] Этот формат соответствовал различным химическим процессам секвенирования, используемым секвенаторами SOLiD. В первоначальных представлениях использовались нуклеотидные основания только в начале последовательности, но более поздние версии включали основания, встроенные через определенные промежутки времени для повышения точности вызова и картирования оснований.
Значения качества для CSFASTQ идентичны значениям для формата Sanger. Инструменты выравнивания различаются предпочтительными вариантами значений качества: некоторые включают показатель качества (установленный на 0, т.е. «!») для ведущего нуклеотида, другие — нет. Архив чтения последовательности включает этот показатель качества.
Эволюция FAST5 и HDF5
[ редактировать ]Формат FAST4 был изобретен как производная от формата FASTQ, в котором каждое из 4 оснований (A,C,G,T) хранило отдельные вероятности. Он был частью Swift basecaller, пакета с открытым исходным кодом для первичного анализа данных последовательностей следующего поколения «от изображений до базовых вызовов».
Формат FAST5 был изобретен как расширение формата FAST4. Файлы FAST5 представляют собой файлы иерархического формата данных 5 (HDF5) со специальной схемой, определенной Oxford Nanopore Technologies (ONT). [ 12 ]
Моделирование
[ редактировать ]Для моделирования чтения FASTQ используется несколько инструментов. [ 13 ] [ 14 ] Сравнение этих инструментов можно увидеть здесь. [ 15 ]
Сжатие
[ редактировать ]Общие компрессоры
[ редактировать ]Инструменты общего назначения, такие как Gzip и bzip2, рассматривают FASTQ как обычный текстовый файл и приводят к неоптимальной степени сжатия. NCBI Архив чтения последовательностей кодирует метаданные с использованием схемы LZ-77. Общие компрессоры FASTQ обычно сжимают отдельные поля (имена чтения, последовательности, комментарии и показатели качества) в файле FASTQ отдельно; к ним относятся DSRC и DSRC2, FQC, LFQC, Fqzcomp и Slimfastq.
Читает
[ редактировать ]Наличие эталонного генома удобно, потому что тогда вместо хранения самих нуклеотидных последовательностей можно просто сопоставить чтения с эталонным геномом и сохранить позиции (указатели) и несоответствия; указатели затем могут быть отсортированы в соответствии с их порядком в ссылочной последовательности и закодированы, например, с помощью кодирования длин серий. Когда покрытие или содержание повторов секвенированного генома велико, это приводит к высокой степени сжатия. В отличие от форматов SAM /BAM, в файлах FASTQ не указан эталонный геном. Компрессоры FASTQ на основе выравнивания поддерживают использование как предоставленного пользователем, так и собранного заново эталонного генома: LW-FQZip использует предоставленный эталонный геном, а Quip, Leon, k-Path и KIC выполняют сборку заново, используя подход на основе графа де Брёйна .
Явное сопоставление чтения и сборка de novo обычно выполняются медленно. Компрессоры FASTQ на основе переупорядочения сначала кластеризуют операции чтения, которые совместно используют длинные подстроки, а затем независимо сжимают операции чтения в каждом кластере после их переупорядочения или сборки в более длинные контиги , достигая, возможно, наилучшего компромисса между временем выполнения и степенью сжатия. SCALCE — первый подобный инструмент, за ним следуют Orcom и Mince. BEETL использует обобщенное преобразование Берроуза-Уиллера для изменения порядка чтения, а HARC достигает более высокой производительности с помощью изменения порядка на основе хэша. Вместо этого AssemblTrie собирает чтения в ссылочные деревья с минимально возможным общим количеством символов в ссылке. [ 16 ] [ 17 ]
Тесты для этих инструментов доступны в . [ 18 ]
Ценности качества
[ редактировать ]Значения качества занимают около половины необходимого дискового пространства в формате FASTQ (до сжатия), поэтому сжатие значений качества позволяет значительно снизить требования к хранению и ускорить анализ и передачу данных секвенирования. В последнее время в литературе рассматриваются как сжатие без потерь, так и сжатие с потерями. Например, алгоритм QualComp [ 19 ] выполняет сжатие с потерями со скоростью (количество бит на значение качества), указанной пользователем. На основе результатов теории искажений скорости он распределяет количество битов таким образом, чтобы минимизировать MSE (среднеквадратическую ошибку) между исходным (несжатым) и восстановленным (после сжатия) значениями качества. Другие алгоритмы сжатия значений качества включают SCALCE. [ 20 ] и Фасткз. [ 21 ] Оба являются алгоритмами сжатия без потерь, которые обеспечивают дополнительный подход к управляемому преобразованию с потерями. Например, SCALCE уменьшает размер алфавита, основываясь на наблюдении, что «соседние» значения качества в целом схожи. Для сравнения см. [ 22 ]
Начиная с HiSeq 2500, Illumina дает возможность выводить качество, которое было крупнозернистым, в ячейки качества. Сгруппированные оценки рассчитываются непосредственно из таблицы эмпирических показателей качества, которая сама привязана к оборудованию, программному обеспечению и химическим препаратам, которые использовались во время эксперимента по секвенированию. [ 23 ]
Расширение файла
[ редактировать ]Для файла FASTQ не существует стандартного расширения , но обычно используются .fq и .fastq.
Конвертеры форматов
[ редактировать ]- Biopython версии 1.51 и более поздних версий (интерконвертирует Sanger, Solexa и Illumina 1.3+)
- EMBOSS версии 6.1.0, патч 1 и выше (объединяет Sanger, Solexa и Illumina 1.3+)
- BioPerl версии 1.6.1 и выше (взаимоконвертирует Sanger, Solexa и Illumina 1.3+)
- BioRuby версии 1.4.0 и выше (взаимно преобразует Sanger, Solexa и Illumina 1.3+)
- BioJava версии 1.7.1 и выше (взаимное преобразование Sanger, Solexa и Illumina 1.3+)
См. также
[ редактировать ]- Формат FASTA , используемый для представления последовательностей генома.
- Форматы SAM и CRAM , используемые для представления считываний секвенатора генома, которые были сопоставлены с последовательностями генома.
- Формат GVF (формат вариаций генома), расширение, основанное на формате GFF3 .
Ссылки
[ редактировать ]- ^ Jump up to: а б Петух, ПИА; Филдс, СиДжей; Гото, Н.; Хойер, МЛ; Райс, премьер-министр (2009). «Формат файла Sanger FASTQ для последовательностей с показателями качества и варианты Solexa/Illumina FASTQ» . Исследования нуклеиновых кислот . 38 (6): 1767–1771. дои : 10.1093/нар/gkp1137 . ПМЦ 2847217 . ПМИД 20015970 .
- ^ Руководство пользователя программного обеспечения для анализа секвенирования: для Pipeline версии 1.4 и CASAVA версии 1.0, от апреля 2009 г. PDF, архивировано 10 июня 2010 г. на Wayback Machine.
- ^ Руководство пользователя программного обеспечения для анализа секвенирования: для конвейерной версии 1.5 и CASAVA версии 1.0, от августа 2009 г. PDF [ мертвая ссылка ]
- ^ Формат карты последовательности/выравнивания, версия 1.0, от августа 2009 г. PDF.
- ↑ Тема скругляка Seqanswer, датированная январем 2011 г., веб-сайт.
- ^ Спецификация формата Elembio AVITI FASTQ https://docs.elembio.io/docs/bases2fastq/outputs/#quality-scores
- ^ Спецификация формата PacBio BAM 10.0.0 https://pacbiofileformats.readthedocs.io/en/10.0/BAM.html#qual
- ^ Руководство по дуплексному базовому вызову Dorado [duplex-tools: использование с Dorado https://github.com/nanoporetech/duplex-tools#usage-with-dorado-recommended ]
- ^ Показатели качества Illumina, Тобиас Манн, Биоинформатика, Сан-Диего, Illumina http://seqanswers.com/forums/showthread.php?t=4721
- ^ Использование анализатора генома Программное обеспечение для управления секвенированием, версия 2.6, номер по каталогу SY-960-2601, номер детали 15009921, ред. A, ноябрь 2009 г. http://watson.nci.nih.gov/solexa/Using_SCSv2.6_15009921_A.pdf [ мертвая ссылка ]
- ^ Сайт проекта SolexaQA
- ^ «Введение_в_Fast5_files» . labs.epi2me.io . Проверено 19 мая 2022 г.
- ^ Хуанг, В; Ли, Л; Майерс, младший; Март, GT (2012). «ART: Симулятор чтения секвенирования нового поколения» . Биоинформатика . 28 (4): 593–4. doi : 10.1093/биоинформатика/btr708 . ПМЦ 3278762 . ПМИД 22199392 .
- ^ Пратас, Д; Пиньо, Эй Джей; Родригес, ЖМ (2014). «XS: Симулятор чтения FASTQ» . Исследовательские заметки BMC . 7:40 . дои : 10.1186/1756-0500-7-40 . ПМЦ 3927261 . ПМИД 24433564 .
- ^ Эскалона, Мерли; Роча, Сара; Посада, Дэвид (2016). «Сравнение инструментов для моделирования данных геномного секвенирования следующего поколения» . Обзоры природы Генетика . 17 (8): 459–69. дои : 10.1038/nrg.2016.57 . ПМК 5224698 . ПМИД 27320129 .
- ^ Гинарт А.А., Хуэй Дж., Жу К., Нуманагич И., Куртад Т.А., Сахинальп СК; и др. (2018). «Оптимальное сжатое представление данных последовательности с высокой пропускной способностью с помощью легкой сборки» . Нат Коммун . 9 (1): 566. Бибкод : 2018NatCo...9..566G . дои : 10.1038/s41467-017-02480-6 . ПМК 5805770 . ПМИД 29422526 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Чжу, Кайюань; Нуманагич, Ибрагим; Сахинальп, С. Дженк (2018). «Сжатие геномных данных». Энциклопедия технологий больших данных . Чам: Международное издательство Springer. стр. 779–783. дои : 10.1007/978-3-319-63962-8_55-1 . ISBN 978-3-319-63962-8 . S2CID 61153904 .
- ^ Нуманагич, Ибрагим; Бонфилд, Джеймс К.; Хач, Фараз; Фогес, Ян; Остерманн, Йорн; Альберти, Клаудио; Маттавелли, Марко; Сахинальп, С. Дженк (24 октября 2016 г.). «Сравнение инструментов высокопроизводительного сжатия данных секвенирования». Природные методы . 13 (12). ООО «Спрингер Сайенс энд Бизнес Медиа»: 1005–1008. дои : 10.1038/nmeth.4037 . ISSN 1548-7091 . ПМИД 27776113 . S2CID 205425373 .
- ^ Очоа, Идоя; Аснани, Химаншу; Бхарадия, Динеш; Чоудхури, Майнак; Вайсман, Цахи; Йона, Голаны (2013). «Qual Comp : новый компрессор с потерями для показателей качества, основанный на теории искажений скорости» . БМК Биоинформатика . 14 :187. дои : 10.1186/1471-2105-14-187 . ПМК 3698011 . ПМИД 23758828 .
- ^ Хач, Ф; Нуманаджик, я; Алкан, К; Сахинальп, Южная Каролина (2012). «SCALCE: усиление алгоритмов сжатия последовательностей с использованием локально согласованного кодирования» . Биоинформатика . 28 (23): 3051–7. doi : 10.1093/биоинформатика/bts593 . ПМК 3509486 . ПМИД 23047557 .
- ^ fastqz. http://mattmahoney.net/dc/fastqz/
- ^ М. Хоссейни, Д. Пратас и А. Пиньо. 2016. Обзор методов сжатия данных биологических последовательностей. Информация 7 (4):(2016): 56
- ^ Техническое примечание Illumina. http://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_understanding_quality_scores.pdf
Внешние ссылки
[ редактировать ]- Веб-страница MAQ , на которой обсуждаются варианты FASTQ