Наследственная диффузная лейкоэнцефалопатия с сфероидами
| Наследственная диффузная лейкоэнцефалопатия с сфероидами (HDL) | |
|---|---|
| Другие имена | Лейкоэнцефалопатия для взрослых с аксональными сфероидами и пигментированной глией, аутосомная доминантная лейкоэнцефалопатия с нейроаксональными сфероидами |
 | |
| Наследственная диффузная лейкоэнцефалопатия с сфероидами наследуется аутосомно -доминантным образом | |
Наследственная диффузная лейкоэнцефалопатия с сфероидами ( HDL ) представляет собой редкое аутосомно -доминантное расстройство взрослого взрослого , характеризующееся головного мозга дегенерацией белого вещества с демиелинизацией и аксональными сфероидами, приводящими к прогрессирующей когнитивной и моторной дисфункции. Сфероиды представляют собой опухоли аксонов с прерывистыми или отсутствием миелиновых оболочек. Считается, что заболевание возникает в результате первичной дисфункции микроглии, которая приводит к вторичному нарушению целостности аксонов, нейроаксональным повреждениям и фокальным аксональным сфероидам, приводящим к демиелинизации . Сфероиды в HDL в некоторой степени напоминают те, которые получают напряжение сдвига в замкнутой травме головы с повреждением аксонов, что приводит к вспышке из -за блокировки аксоплазматического переноса . В дополнение к травме, аксональные сфероиды можно найти в старом мозге, инсультах и при других дегенеративных заболеваниях. [ 1 ] У HDL неясно, происходит ли демиелинизация до аксонов сфероидов или что запускает нейродегенерацию после, по -видимому, нормальное развитие мозга и белого вещества, хотя генетический дефицит предполагает, что демиелинизация и патология аксонов могут быть вторичными по отношению к дисфункции микроглиального. [ 2 ] Клинический синдром у пациентов с HDLS не является специфическим, и его можно принять при болезни Альцгеймера , лобно -височной деменции , атипичного паркинсонизма , рассеянного склероза или дегенерации кортикобаза . [ 3 ]
Симптомы
[ редактировать ]С симптомами изменений личности, поведенческих изменений, деменции , депрессии и эпилепсии, HDL обычно неправильно диагностируются для ряда других заболеваний. [ 4 ] Например, деменция или лобно -височные изменения поведения обычно привели к тому, что некоторые клиницисты по ошибке рассматривают диагнозы, такие как болезнь Альцгеймера, лобно -височная деменция или нетипичный паркинсонизм. Наличие изменений белого вещества привело к неправильной диагностике рассеянного склероза. HDL обычно проявляются с нейропсихиатрическими симптомами, прогрессирующими до деменции и через несколько лет показывают моторную дисфункцию. В конечном итоге пациенты становятся зависимыми от инвалидных колясок. [ 3 ]
Дегенерация белого вещества связана с дифференциальными диагнозами из других лейкодистрофий, таких как метахроматическая лейкодистрофия (MLD), болезнь Краббе (лейкодистрофия глобоидных клеток) и X-связанная адренолекодистрофия (X-ADL). [ 2 ]
| Болезнь | Эксклюзивная черта |
|---|---|
| Миллиард | Накопление метахроматического материала в белом веществе |
| Крабовая болезнь | Наличие глобоидных клеток, полученных из микроглии, которые имеют множественные ядра |
| X-ald | Преобладающая аномалия белого вещества с темами |
| Исчезающая болезнь белого вещества (VWM) |
|
| Насу-Хакола |
|
Нейропсихиатрические симптомы
[ редактировать ]Многие нейропсихиатрические симптомы были идентифицированы в клинических исследованиях пациентов с HDLS. К ним относятся тяжелая депрессия и тревога, которые были выявлены примерно в 70% семей HDLS, граничащие с суицидальными тенденциями и злоупотреблением психоактивными веществами, такими как алкоголизм . Кроме того, пациенты могут проявлять дезориентацию, растерянность, возбудимость, раздражительность, агрессивность, измененное психическое состояние, потерю способности выполнять обученные движения ( апраксия ) или неспособность говорить ( мутизм ). [ 3 ]
Моторное нарушение
[ редактировать ]Люди с ЛПВП могут развивать тремор, уменьшение движения тела, неустойчивость ( паркинсонизм , мышцы на одной стороне тела в постоянном сокращении ( спастический гемипарез ), нарушение моторной и сенсорной функции в нижних конечностях ( парапарезис ), паралич, что приводит к частичному или общему Потеря всех конечностей и туловища ( тетрапарезис ) и отсутствие добровольной координации мышечных движений ( атаксия ). [ 3 ]
Причины
[ редактировать ]Причиной ЛПВП в большинстве семейств является мутация в рецепторе стимулирующего фактора 1 колонии (CSF1R), фактор роста для микроглии и моноцитов/макрофагов, что позволяет предположить, что дисфункция микроглии может быть первичной в HDL. [ 4 ]
Мутации сосредоточены в тирозинкиназы домене (TKD) белка. Мутации были в основном обнаружены в экзонах 12-22 внутриклеточного TKD , в том числе 10 миссенс-мутаций , которые имеют единую удаление нуклеотидов и делецию единого кодона, которая состоит из триплета нуклеотидов, которые были удалены, вызывая целую аминокислоту не кодироваться. три мутации сайта сплайсинга Кроме того, были идентифицированы в рамке , которые вызывали делецию экзона , экспрессированную нуклеотидную последовательность, что приводило к удалению более 40 аминокислот в ТКД. [ 4 ]
Это определение основывалось на генетических исследованиях 14 семейств HDLS, подтверждающих мутации в этом гене. Белок рецептора CSF1 в основном функционирует в регуляции, выживании, пролиферации и дифференцировке микроглиальных клеток. [ 5 ] Механизм дисфункции микроглии из -за мутаций в CSF1R с потерей миелина и образования аксонов аксонов остается неизвестным. Дальнейшие исследования необходимы для лучшего понимания патогенеза заболевания .
Патология
[ редактировать ]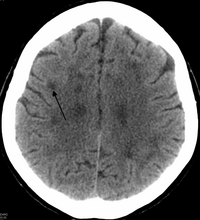
У HDLS происходит увеличение боковых желудочков и отмеченное истончение или ослабление белого мозга. [ 6 ] Потеря белого вещества вызвана потерей миелина . Эти изменения связаны с диффузным глиозом , умеренной потерей аксонов и многих аксонов. [ 1 ]
Активированная или амебоидная микроглия и макрофаги, которые содержат мусор миелина, липидные капли и коричневые автофлуоресцентные пигментные гранулы, обнаружены в областях с демиелинизацией и аксональными сфероидами. В сильно дегенерированных областях есть много больших реактивных астроцитов, заполненных глиальными фибрилл . [ 1 ]
В случаях вскрытия было показано, что аномалии белого вещества относительно ограничены головным мозгом , избегая при этом мозжечка и многие основные волокнистые тракты нервной системы. Исключением являются кортикоспинальные тракты (пирамидальные тракты) в стволе мозга , а иногда и в спинном мозге . [ 2 ]
Патология мозга HDL напоминает патологическую болезнь Насу-Хакола (поликистозная липолемембрановая остеодисплазия с склерозирующей лейкоэнцефалопатией). [ 7 ]
Диагноз
[ редактировать ]Исследования по состоянию на 2012 год включают исследования микроглии. Эта работа дополнительно прояснит, является ли заболевание в первую очередь дефектом в функции микроглии. Для такого исследования микроглиальные клетки из HDLS Kindred могут быть культивированы из мозга вскрытия и проанализированы по сравнению с нормальными микроглиальными клетками на основе различий в возникновениях мутаций и экспрессии фактора роста. [ 5 ]
Дифференциальный диагноз
[ редактировать ]Связанные расстройства в том же спектре заболевания, что и ЛПВП, включают болезнь Насу-Хакола ( поликистозная липоомембрановая остеодисплазия с склерозирующей лейкоэнцефалопатией ), и тип лейкодистрофии с пигментированными макрофагами, называемым пигментальной ортохроматической лейкодизстрофией (Pold). [ 3 ] В дополнение к заболеванию белого вещества, Насу-Хакола вызывает кисты костей. Это вызвано мутациями в генах, участвующих в одном и том же колонии, стимулирующем факторе (CSF) каскада, как идентифицировано в HDL. [ 8 ]
Болезнь Насу-Хакола, по-видимому, вызвана мутациями в тирозинки-связывающем белке тирозинкиназы ( Tyrobp -также известный как DAP12) или запускающий рецептор, экспрессируемый на белке миелоидных клеток 2 ( TREM2 ). В то время как различные генные мутации происходят в пути для Насу-Хакола и HDL, оба характеризуются дегенерацией белого вещества с помощью аксонов сфероидов. Нынешние исследователи в этой области считают, что более глубокий анализ и сравнение двух генетических нарушений при этих расстройствах могут привести к лучшему пониманию механизмов заболевания при этих редких расстройствах. У POLD демонстрирует невоспаливающая демиелинизация аксонов с начальными симптомами эйфории, апатии, головной боли и руководителей . В то время как HDLS является аутосомно -доминантным, некоторые семьи с POLD имеют особенности, которые предполагают аутосомно -рецессивное наследование. [ 9 ] Тем не менее, недавно было показано, что POLD имеет такую же генетическую основу, что и HDL.
Клинические и генеалогические исследования
[ редактировать ]Чтобы лучше понять заболевание, исследователи ретроспективно рассмотрели медицинские записи пробандов и других, которые были оценены с помощью клинических осмотров или анкет. Образцы крови собираются из семейств пробандов для генетического тестирования . Эти члены семьи оцениваются с использованием их стандартной истории болезни , о их прогрессировании симптомов Паркинсона ( Объединенная шкала оценки болезни Паркинсона ) и о их прогрессировании когнитивных нарушений, таких как деменция ( тест Фольштейна ). [ 2 ]
Нейровизуализация
[ редактировать ]Стандартные МРТ были выполнены на 1,5 сканерах Tesla с толщиной 5 мм и расстоянием 5 мм для скрининга поражений белого вещества в идентифицированных семействах. Если интенсивность сигналов МРТ -сканирования выше в областях белого вещества, чем в областях серого вещества, считается, что пациент подвергается риску ЖПП, хотя ряд других расстройств также могут вызывать изменения белого вещества, а результаты не являются диагностическими без генетических тестирование или патологическое подтверждение. [ 2 ]
Патология
[ редактировать ]Срезы ткани из биопсии головного мозга или мозга вскрытия обычно встроены в парафин, из которого срезы разрезаются и устанавливаются на стеклянных предметных стеклах для гистологических исследований. Специальные пятна для миелина и патологии аксонов показывают аномальные изменения, которые характерны для HDL, идентифицированы в белом веществе неокортекс , базальных ганглиев , таламуса , среднего мозга , понков и спинного мозга. [ 2 ] [ 10 ] В дополнение к обычным гистологическим методам ( окрашивание H & E ) образцы оцениваются с помощью иммуногистохимии для убиквитина , белка -предшественника амилоида и нейрофиламента для характеристики изменений аксонов и основного белка миелина для патологии миелина. Иммуногистохимические пятна для микроглии (CD68 или HLA-DR) и астроцитов (GFAP) также являются полезными методами для характеристики патологии белого вещества. [ 6 ] С аналогичной патологией с POLD, HDLS обычно сгруппируется как лейкоэнцефалопатия для взрослых с аксональными сфероидами и пигментированным глией (ALSP), чтобы привлекать их индивидуально недооцененные условия, усиливающие внимание. [ 3 ]
Классификация
[ редактировать ]HDLs подпадает под категорию заболеваний белого мозга, называемых лейкоэнцефалопатиями, которые характеризуются некоторой степенью дисфункции белого вещества. HDLS имеет поражения белого вещества с аномалиями в миелиновой оболочке вокруг аксонов, где причинные влияния постоянно изучаются на основе недавних генетических результатов. Исследования Sundal и коллег из Швеции показали, что аллель риска у кавказцев может быть причинным, поскольку выявленные случаи до сих пор были среди крупных кавказских семей. [ 2 ]
Управление
[ редактировать ]Этот раздел пуст. Вы можете помочь, добавив к этому . ( Октябрь 2017 г. ) |
Эпидемиология
[ редактировать ]Средний клинический профиль из опубликованных исследований показывает, что средний возраст начала для пациентов с HDL составляет 44,3 года со средней продолжительностью заболевания 5,8 года и средним возрастом смерти в 53,2 года. [ 2 ] [ 11 ] По состоянию на 2012 год было выявлено около 15 случаев, по меньшей мере, с 11 спорадическими случаями HDL. [ 2 ] [ 11 ] Случаи HDLS были расположены в Германии, Норвегии, Швеции и Соединенных Штатах, демонстрируя международное распределение, сосредоточенное на северной Европе и Соединенных Штатах. [ 2 ]
Благодаря изучению многочисленных родственников было обнаружено, что заболевание не возникало среди только мужчин или женщин, а скорее была равномерно распределена, что свидетельствует о аутосомно, а не для генетического расстройства, связанного с полом . Также было отмечено, что случаи HDLS не пропускали поколения, как это произойдет с рецессивным наследством, и, как таковое, было названо аутосомно -доминантом. [ 2 ]
История
[ редактировать ]Это заболевание было впервые описано в 1984 году Axelsson et al. В большой шведской родословной. [ 12 ] Это расстройство, более известное невропатологам, чем врачи. Невропатолог с интересом к HDLS, доктор Деннис В. Диксон, выявил ряд случаев из исследования невропатологии мозга, представленных для исследования семейной деменции и нарушений движения в Нью-Йорке, а затем во Флориде. Признание важности этого расстройства в качестве причины деменции взрослых и расстройств движения было дополнительно повышено в 1997 году в клинике Майо , когда доктор Збигьют К. Весзолек идентифицировал семью с ЛПВП, которая изначально считалась из -за другого заболевания (процесс (процесс ( FTDP-17), но только вскрытие одного, а затем другие члены семьи показали, что это HDL. В 2005 году WSzolek создал международный консорциум для выявления других семей и для сбора образцов ДНК или мозга от членов семьи для невропатологического подтверждения и генетических исследований в клинике Майо во Флориде. [ 2 ]
Смотрите также
[ редактировать ]- Нейродегенерация
- Лейкоэнцефалопатия с исчезающим белым веществом
- Лейкоэнцефалопатия с нейроаксональными сфероидами
- Микроцефалия
Ссылки
[ редактировать ]- ^ Подпрыгнуть до: а беременный в Lin, WL, Wszolek, Zk, & Dickson, DW (2010). Наследственная диффузная лейкоэнцефалопатия с сфероидами: ультраструктурные и иммуноэлектронные микроскопические исследования. Int J Clin Exp Pathol, 3 (7), 665-674.
- ^ Подпрыгнуть до: а беременный в дюймовый и фон глин час я Дж k л м Sundal, C., Lash, J., Aasly, J., Oygarden, S., Roeber, S., Kretschman, H.,. Полем Полем Wszolek, ZK (2012). Наследственная диффузная лейкоэнцефалопатия с аксональными сфероидами (HDL): неправильно диагностированная болезнь. J Neurol Sci, 314 (1-2), 130-137. Doi : 10.1016/j.jns.2011.10.006
- ^ Подпрыгнуть до: а беременный в дюймовый и фон Wider, C., Van Gerpen, JA, Dearmond, S., Shuster, EA, Dickson, DW, & Wszolek, ZK (2009). Лейкоэнцефалопатия с сфероидами (HDL) и пигментальной лейкодистрофией (POLD): единственная сущность? Неврология, 72 (22), 1953–1959. Doi : 10.1212/wnl.0b013e3181a826c0
- ^ Подпрыгнуть до: а беременный в Rademakers, R., Baker, M., Nicholson, A., Rutherford, N., Finch, N., Soto-ortolaza, A.,. Полем Полем Wszolek, Z. (2012). Мутации в рецепторе стимулирующего фактора 1 колонии (CSF1R) вызывают наследственную диффузную лейкоэнцефалопатию с сфероидами. Нарушения движения, 27, S399-S400.
- ^ Подпрыгнуть до: а беременный Kinoshita, M., Yoshida, K., Oyanagi, K., Hashimoto, T. & Ikeda, S. (2012). Наследственная диффузная лейкоэнцефалопатия с аксональными сфероидами, вызванными мутацией R782H в CSF1R: отчет о случае. Журнал неврологических наук, 318 (1-2), 115-118. Doi : 10.1016/j.jns.2012.03.012
- ^ Подпрыгнуть до: а беременный Baba, Y., Ghetti, B., Baker, MC, Uitti, RJ, Hutton, ML, Yamaguchi, K.,. Полем Полем Wszolek, ZK (2006). Наследственная диффузная лейкоэнцефалопатия с сфероидами: клинические, патологические и генетические исследования нового родственника. Acta Neuropathol, 111 (4), 300-311. Два : 10.1007/s00401-006-0046-z
- ^ Hancock, N., Poon, M., Taylor, B. & McLean, C. (2003). Наследственная диффузная лейкоэнцефалопатия с сфероидами. J Neurol Neurosurg Psychiatry, 74 (9), 1345–1347.
- ^ Paloneva, J., Mandelin, J., Kiialainen, A., Böhling, T., Prudlo, J., Hakola, P.,. Полем Полем Пелтонен Л. (2003). Дефицит DAP12/TREM2 приводит к нарушению дифференцировки остеокластов и остеопоротических характеристик. Журнал экспериментальной медицины, 198 (4), 669-675.
- ^ Knaap, Marjo S. & Valk, Jaap. (2005). Пигментная ортохромная лейкодистрофия Магнитный резонанс миелининации и миелиновых расстройств (стр. 557-558): Springer Berlin Heidelberg.
- ^ Van Gerpen, JA, Wider, C., Broderick, DF, Dickson, DW, Brown, LA, & Wszolek, ZK (2008). Понимание динамики наследственной диффузной лейкоэнцефалопатии с аксональными сфероидами. Неврология, 71 (12), 925-929. Два : 10.1212/01.wnl.0000325916.30701.21
- ^ Подпрыгнуть до: а беременный Sundal, C., van Gerpen, JA, Nicholson, AM, Wider, C., Shuster, EA, Aasly, J.,. Полем Полем Wszolek, ZK (2012). Характеристики МРТ и оценка в ЖНП из -за мутаций генов CSF1R. Неврология, 79 (6), 566-574. Doi : 10.1212/wnl.0b013e318263575a
- ^ Axelsson, R., Roytta, M., Sourander, P., Akesson, Ho, & Andersen, O. (1984). Наследственная диффузная лейкоэнцефалопатия с сфероидами. Acta Psychiatr Scand Suppl , 314, 1-65.