Лейкодистрофия
| Лейкодистрофия | |
|---|---|
 | |
| Т2-взвешенное аксиальное сканирование человеческого мозга на уровне хвостатых головок демонстрирует выраженную потерю заднего белого вещества с уменьшенным объемом и повышенной интенсивностью сигнала. Переднее белое вещество сохранено. Характеристики соответствуют Х-сцепленной адренолейкодистрофии . | |
| Специальность | Неврология |
Лейкодистрофии представляют собой группу обычно наследственных заболеваний, характеризующихся дегенерацией белого вещества головного мозга. [1] Слово лейкодистрофия происходит от греческих корней leuko — «белый», Dys — «ненормальный» и trop — «рост». Лейкодистрофии вызваны несовершенным ростом или развитием глиальных клеток , которые образуют миелиновую оболочку , жировое изолирующее покрытие вокруг нервных волокон . [2] Лейкодистрофии можно классифицировать как гипомиелинизирующие или демиелинизирующие заболевания соответственно, в зависимости от того, присутствует ли повреждение до рождения или возникает после. Хотя все лейкодистрофии являются результатом генетических мутаций, [3] другие демиелинизирующие заболевания имеют аутоиммунную , инфекционную или метаболическую этиологию. [4]
Когда происходит повреждение белого вещества, последующие иммунные реакции могут привести к воспалению в центральной нервной системе (ЦНС) наряду с потерей миелина. Дегенерацию белого вещества можно увидеть на МРТ , которая используется для диагностики лейкодистрофии. Лейкодистрофия характеризуется специфическими симптомами, включая снижение двигательной функции, мышечную ригидность и возможное ухудшение зрения и слуха. Хотя заболевание смертельно, возраст начала является ключевым фактором, поскольку типичная продолжительность жизни младенцев составляет 2–8 лет, тогда как взрослые обычно живут более десяти лет после начала заболевания. Варианты лечения ограничены, хотя трансплантация гемопоэтических стволовых клеток с использованием костного мозга или пуповинной крови , по-видимому, помогает при определенных типах лейкодистрофии, в то время как проводятся дальнейшие исследования.
Совокупная заболеваемость лейкодистрофиями оценивается в 1 на 7600. [5] Большинство типов связаны с наследованием Х-сцепленного рецессивного или Х-сцепленного доминантного признака, в то время как другие, хотя и включают дефектный ген, являются результатом спонтанной мутации, а не генетического наследования .
Симптомы и признаки
[ редактировать ]Некоторые специфические симптомы варьируются от одного типа лейкодистрофии к другому, но подавляющее большинство симптомов являются общими, поскольку причины заболевания обычно имеют одинаковые последствия. Симптомы зависят от возраста начала заболевания, которое преимущественно приходится на младенчество и ранний детский возраст, хотя точное время начала может быть трудно определить. Распространены повышенная раздражительность и повышенная чувствительность к окружающей среде, а также некоторые характерные физические признаки, включая ригидность мышц и наклоненную назад голову. [6] Ботокс-терапия часто используется для лечения пациентов со спастичностью. [7] У подростков и взрослых симптомы проявляются сходными, включая снижение или потерю слуха и зрения. Хотя у детей действительно наблюдаются оптические и слуховые дегенерации, течение болезни обычно слишком быстрое, что приводит к относительно быстрой смерти, тогда как взрослые могут жить с этими состояниями в течение многих лет. У детей спастическая активность часто предшествует прогрессирующей атаксии и быстрому ухудшению когнитивных функций, которое описывается как умственная отсталость . [8] Эпилепсия является обычным явлением для пациентов всех возрастов. [9] У более прогрессирующих пациентов наблюдается слабость глотания , что приводит к приступам спастического кашля из-за вдыхания слюны. Классическое симптоматическое прогрессирование ювенильной Х-сцепленной адренолейкодистрофии показано в фильме 1992 года « Масло Лоренцо» . [10]
Течение и график зависят от возраста начала заболевания: у младенцев продолжительность жизни составляет 2–8 лет, у подростков – 2–10 лет, а у взрослых – обычно 10+ лет. Взрослые обычно наблюдают длительный период стабильности, за которым следует снижение до вегетативного состояния и смерть. [6] Хотя методы лечения существуют, большинство из них находятся на экспериментальной стадии и могут лишь обещать остановку прогрессирования симптомов, хотя некоторые методы генной терапии показали некоторое симптоматическое улучшение. [11] Изнурительное течение болезни привело к многочисленным философским и этическим спорам по поводу экспериментальных клинических испытаний, прав пациентов и самоубийства с помощью врача . [12]
Причины
[ редактировать ]Хотя более конкретные причины лейкодистрофии зависят от типа, существуют общие патофизиологические закономерности, которые можно наблюдать среди всех типов. Прежде всего, лейкодистрофия — это нейродегенеративное заболевание, которое всегда является результатом как нарушения, так и сохранения миелиновых оболочек, окружающих аксоны нейронов в центральной нервной системе, в результате генетической мутации . [13] Миелин представляет собой жирное белое вещество, которое действует как электрический изолятор и покрывает аксоны, ускоряя импульсы (то есть потенциалы действия ), распространяющиеся по аксону. Таким образом, естественным результатом потери этого вещества является снижение эффективности распространения импульсов. Поскольку миелин вырабатывается олигодендроцитами (типом глиальных клеток ) в центральной нервной системе, причину легко искать в мутации или неисправности этих клеток и других глиальных клеток. [ нужна ссылка ]
Генетическое влияние
[ редактировать ]
Наследственные формы лейкодистрофии обычно являются результатом аутосомно-рецессивного типа наследования, хотя нередки случаи доминантного наследования, как в случае лейкодистрофии с началом во взрослом возрасте. [14] Это означает, что пораженный аллель переносится на аутосомной , или неполовой, хромосоме и маскируется доминантным, незатронутым фенотипом . Другими словами, чтобы человек унаследовал фенотип лейкодистрофии, он или она должны нести два рецессивных мутантных аллеля. болезнь Краббе и метахроматическая лейкодистрофия К таким типам относятся (МЛД). MLD обнаружен на 22 хромосоме человека в положении q13.31. [15] Другим типом наследственной лейкодистрофии является Х-сцепленная адренолейкодистрофия (Х-АЛД). Как следует из названия, этот тип лейкодистрофии является результатом мутации, обнаруженной в Х-хромосоме . Он также передается по рецессивному типу. Х-хромосома является половой хромосомой , и поскольку у женщин есть два «шанса» на приобретение нормальной Х-хромосомы (один материнский х, один отцовский), а у мужчин только один шанс (один материнский х), это заболевание с большей вероятностью наблюдается у мужчин, чем у женщин. Мутация, приводящая к лейкодистрофии у взрослых, картирована в 5q23. [14]
Патофизиология
[ редактировать ]Хотя существует около 40 различных типов лейкодистрофии, многие из них не имеют официальных и всесторонних исследований. Большая часть исследований на данный момент была проведена по пяти типам: (1) метахроматическая лейкодистрофия (МЛД), (2) болезнь Краббе , (3) Х-сцепленная адренолейкодистрофия (АЛД), (4) болезнь Канавана и (5) болезнь Александера. болезнь . Каждый тип лейкодистрофии имеет уникальную патофизиологию , но все пять из них тем или иным образом влияют на подмножество глиальных клеток, тем самым нарушая выработку и поддержание миелина, и обычно включают мутацию, затрагивающую гены, которые кодируют ферменты, необходимые для катаболизма очень длинноцепочечных клеток. жирные кислоты (VLCFA), которые токсичны для миелин-продуцирующих клеток центральной нервной системы. [16]
Метахроматическая лейкодистрофия
[ редактировать ]Метахроматическая лейкодистрофия является результатом генетических дефектов ферментов, связанных с клеточным компартментом, называемым лизосомой . MLD — одна из двух лейкодистофий, которые также являются лизосомальным заболеванием накопления . MLD наследуется аутосомно -рецессивным результатом мутаций трех различных аллелей ARSA , которые кодируют фермент арилсульфатазу А (ASA или иногда ARSA), также называемую сульфатидсульфатазой путем и является . [17] АСК отвечает за расщепление сульфатидов и сфинголипидов, присутствующих в мембранах нейронов, а также в миелине. Когда происходит мутация в гене, кодирующем АСК, снижается выработка АСК, что впоследствии приводит к уменьшению деградации сульфатидов, вызывая тем самым их накопление. [17] Такое накопление сульфатидов токсично для олигодендроцитов, миелин-продуцирующих клеток ЦНС, что эффективно приводит к нарушению структуры миелина с последующей демиелинизацией . Характер наследования трех различных аллелей влияет на то, какой тип MLD развивается у человека. Два нулевых аллеля ответственны за инфантильную версию и не позволяют производить ASA. У гетерозиготного человека (один нулевой аллель, один ненулевой аллель) развивается ювенильная форма и наблюдается некоторое производство ASA, тогда как у человека с двумя мутированными ненулевыми аллелями развивается взрослая форма. [18]
Крабовая болезнь
[ редактировать ]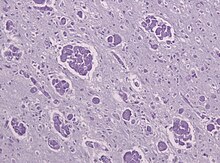
Как и МЛД, болезнь Краббе — это еще один тип лейкодистрофии с аутосомно-рецессивным наследованием, являющийся результатом лизосомального нарушения накопления . Это происходит из-за делеции экзона 16 гена GALC , которая вызывает мутацию сдвига рамки считывания, приводящую к преждевременному образованию стоп-кодона . Ген GALC, обнаруженный на хромосоме 14 в положении 31 (14q31), кодирует фермент бета-галактоцереброзидазу (GALC). [19] GALC — лизосомальный фермент, ответственный за катаболизм галактолипидов , особенно токсичного липида психозина , которые широко распространены по всему мозгу. Таким образом, дефицит GALC вызывает накопление этих жирных кислот , что приводит к вторжению клеток, называемых «глобоидными макрофагами », которые разрушают олигодендроциты, тем самым подавляя любое дальнейшее образование миелина. [20] Учитывая наличие глобоидных макрофагов, сгруппированных вблизи белого вещества , болезнь Краббе часто называют глобоидноклеточной лейкодистрофией.
болезнь Канавана
[ редактировать ]Болезнь Канавана — менее изученный тип лейкодистрофии, который, как и МЛД и болезнь Краббе, также наследуется по аутосомно-рецессивному типу. Это связано с мутацией гена ASPA , который кодирует аспартоацилазу , фермент, необходимый для метаболизма N-ацетил-L-аспартата (NAA). Мутация вызывает дефицит аспартоациклазы. НУК участвует в образовании липидов ; если он не расщепляется аспартоацилазой, уровень липидов в мозге увеличивается, вызывая демиелинизацию. [21]
Х-сцепленная адренолейкодистрофия
[ редактировать ]При Х-сцепленной адренолейкодистрофии (X-ALD) происходит мутация в пероксисомальной АТФ-связывающей кассете ( транспортер ABC ). Это приводит к воспалительной демиелинизации головного мозга , вызванной дестабилизацией миелина. [22] Воспалительная демиелинизация начинается в мозолистом теле и медленно распространяется наружу в оба полушария. У пациентов с Х-АЛД аномально высокие уровни ЖКОДЦ накапливаются в различных тканях и жидкостях организма. Эта повышенная концентрация затем включается в различные сложные липиды, в которых ЖКЖК обычно не обнаруживаются. [22] Было обнаружено, что это напрямую связано с церебральным воспалением X-ALD. Предполагается, что накопленные и встроенные ЖКОДЦ в сложные липиды могут приводить к дестабилизации миелиновой оболочки и, в конечном итоге, к демиелинизации. [ нужна ссылка ]
болезнь Александра
[ редактировать ]Болезнь Александера отличается от упомянутых выше лейкодистрофий тем, что является результатом спонтанной мутации , то есть не передается по наследству. Мутация, обнаруженная у больного человека, не обнаруживается ни у его родителей. Симптомы возникают в результате накопления глиального фибриллярного кислого белка (GFAP) в результате мутации гена GFAP , белок которого не находится в ассоциации с лизосомами или пероксисомами, а представляет собой промежуточную нить, связанную с ядерной оболочкой . [23] Промежуточные филаменты представляют собой белки, ответственные за состав клеточного цитоскелета ; таким образом, этот тип мутации вызывает аномальное структурное развитие клеток человека. цитоскелета и молекул-транспортеров наблюдались дефекты В астроцитах больных людей . Эти астроциты содержат аномально высокие уровни белка GFAP, что влияет на их развитие и функцию. [24]
Диагностика
[ редактировать ]Дегенерацию белого вещества , отражающую дегенерацию миелина, можно увидеть при базовой МРТ и использовать для диагностики лейкодистрофий всех типов. взвешенные изображения T-1 и T-2 с инверсионным восстановлением с жидкостным ослаблением (FLAIR). Наиболее часто используемый подход — [25] Также могут быть проведены электрофизиологические и другие виды лабораторных исследований. В частности, скорость нервной проводимости учитывают, чтобы отличить лейкодистрофию от других демиелинизирующих заболеваний , а также различать отдельные лейкодистрофии. Например, люди с X-ALD имеют нормальную скорость проводимости, тогда как люди с болезнью Краббе или метахроматической лейкодистрофией имеют отклонения в скорости проводимости. [25] Панели мультигенного секвенирования недифференцированной лейкодистрофии предлагаются для быстрой молекулярной диагностики после генетического консультирования. [ нужна ссылка ]
Типы
[ редактировать ]К конкретным типам лейкодистрофии относятся следующие с соответствующими кодами МКБ-10, если таковые имеются: [ нужна ссылка ]
- (E75.2) Болезнь Александера
- (E75.2) Болезнь Канавана
- (E75.2) Гипомиелинирующая лейкодистрофия 7 типа (синдром 4H)
- (E75.2) Болезнь крабов
- (E75.2) Метахроматическая лейкодистрофия
- (E75.2) Болезнь Пелицеуса–Мерцбахера
- (E75.5) Церебротендинальный ксантоматоз
Уход
[ редактировать ]При многих различных типах лейкодистрофии, имеющих множество причин, методы лечения будут различаться для каждого типа. Исследования и клинические испытания направлены на поиск методов лечения каждой из различных лейкодистрофий. Трансплантация стволовых клеток и генная терапия кажутся наиболее многообещающими методами лечения всех лейкодистрофий, при условии, что они проводятся как можно раньше, до обширного неврологического повреждения.
В отношении гипомиелинизирующих лейкодистрофий многообещающими кажутся терапевтические исследования клеточной терапии. Клетки-предшественники олигодендроцитов и нейральные стволовые клетки были успешно трансплантированы и год спустя оказались здоровыми. Карты фракционной анизотропии и радиальной диффузии показали возможную миелинизацию в области трансплантата. [26] [ нужно обновить ] Индуцированные плюрипотентные стволовые клетки , клетки-предшественники олигодендроцитов, коррекция генов и трансплантация, способствующие созреванию, выживанию и миелинизации олигодендроцитов, по-видимому, являются основными путями возможного лечения. [26] [ нужно обновить ]
Для трех типов лейкодистрофии ( Х-сцепленная адренолейкодистрофия (X-ALD), метахроматическая лейкодистрофия (MLD) и болезнь Краббе (глобоидно-клеточная лейкодистрофия - GLD) генная терапия с использованием аутологичных гемопоэтических стволовых клеток для переноса здоровой копии гена, вызывающего заболевание. Было показано, что с лентивирусными векторами успех достигается, и он использовался в клинических испытаниях X-ALD и MLD. [11] [27] Было показано, что прогрессирование X-ALD прерывается генной терапией гемопоэтическими стволовыми клетками, хотя проксимальная причина остановки демиелинизации и количество необходимых стволовых клеток неясны. [11] Хотя в мозге продолжает происходить накопление жирных кислот с очень длинной цепью , это, по-видимому, не является непосредственным причинным фактором заболевания, поскольку генная терапия не корректирует накопление. [11] [ нужно обновить ]
Для тех лейкодистрофий, которые возникают в результате дефицита ферментов лизоцима, таких как болезнь Краббе , заместительная ферментная терапия кажется многообещающей. Однако доставка ферментов оказывается затруднительной, поскольку гематоэнцефалический барьер серьезно ограничивает то, что может проникнуть в центральную нервную систему. [11] Текущие исследования генной терапии метахроматической лейкодистрофии были рассмотрены с упором на трансплантацию ex vivo генетически модифицированных гемопоэтических стволовых клеток. [28] [ нужно обновить ]
Эпидемиология
[ редактировать ]
В настоящее время ни одно исследование не выявило более высокой распространенности большинства типов лейкодистрофии в каком-либо одном месте в мире. Однако среди еврейского населения распространенность болезни Канавана выше. Каждый 40 человек еврейского происхождения ашкенази является носителем болезни Канавана. [29] Это экстраполируется примерно на 2,5%. Кроме того, из-за аутосомно-рецессивного типа наследования не обнаружено существенных различий между мужчинами и женщинами для большинства типов лейкодистрофии, включая, помимо прочего, метахроматическую лейкодистрофию, болезнь Краббе, болезнь Канавана и болезнь Александера. Единственным исключением из этого правила является любой тип лейкодистрофии, переносимый на половой хромосоме , например Х-сцепленная адренолейкодистрофия, которая передается на Х-хромосоме. Из-за характера наследования Х-сцепленных заболеваний этим типом лейкодистрофии чаще страдают мужчины, в то время как женщины-носители часто страдают симптомами, хотя и не так тяжело, как мужчины. [30]
Исследовать
[ редактировать ]Национальный институт неврологических расстройств и инсульта (NINDS при Национальном институте здравоохранения США ) поддерживает исследования генетических нарушений, включая лейкодистрофии. [31] NINDS также поддерживает исследователей, которые работают с Сетью клинических исследований Глобальной инициативы по лейкодистрофии (GLIA-CTN), которая способствует достижениям в диагностике и лечении лейкодистрофий. [32]
Европейская ассоциация лейкодистрофии также поддерживает исследования лейкодистрофии. По состоянию на 2020 год профинансировано более 387 исследовательских проектов. Каждый год ELA приглашает международное научное сообщество представить исследовательские проекты в области генетических лейкодистрофий, белого вещества головного мозга у недоношенных детей и восстановления миелина. [33]
Общество
[ редактировать ]Объединенный фонд лейкодистрофии (ULF), основанный в 1982 году, является некоммерческой добровольной медицинской организацией, занимающейся финансированием передовых исследований и предоставлением пациентам и их семьям информации о заболеваниях и направлений к врачу. [34]
Cure MLD — это глобальная сеть защитников прав пациентов и некоммерческих организаций, призванная помогать семьям, пострадавшим от метахроматической лейкодистрофии (MLD). [35]
Фонд MLD был основан Дином и Терин Зур в 2001 году после того, как в 1995 году двум их дочерям был поставлен диагноз MLD. Фонд MLD обслуживает семьи и работает с исследователями, врачами, регулирующими органами, плательщиками и политиками по всему миру по вопросам MLD, лейкодистрофии, лизосомальных и редких заболеваний. [36]
Альянс по лейкодистрофии работает над повышением осведомленности и качества помощи людям с лейкодистрофией. [37]
Джилл Келли и ее муж, НФЛ защитник Джим Келли , основали Hunter's Hope Foundation для финансирования исследований после того, как у их сына Хантера (1997–2005) была диагностирована детская лейкодистрофия Краббе. [38]
Мэтью и Майкл Кларк из Халла , Великобритания, страдали этим заболеванием. Оба умерли в 2013 и 2016 годах соответственно. [ нужна ссылка ] Их история стала темой документального фильма Channel 4 «Загадочная история братьев Кларк» . [39] [40]
Аугусто и Микаэла Одоне основали проект «Миелин» после того, как их сыну Лоренцо поставили диагноз адренолейкодистрофия (АЛД). Фильм 1992 года « Масло Лоренцо» представляет собой правдивую историю о мальчике, страдающем адренолейкодистрофией (АЛД). [ нужна ссылка ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Сачдев, Перминдер С.; Кешаван, Матчери С. (15 марта 2010 г.). Вторичная шизофрения . Издательство Кембриджского университета. стр. 241–. ISBN 978-0-521-85697-3 . Проверено 15 августа 2011 г.
- ^
 Одно или несколько предыдущих предложений включают текст из этого источника, который находится в свободном доступе : « Информационная страница лейкодистрофии ». Национальный институт неврологических расстройств и инсульта. 25 мая 2017 г. Проверено 18 марта 2018 г.
Одно или несколько предыдущих предложений включают текст из этого источника, который находится в свободном доступе : « Информационная страница лейкодистрофии ». Национальный институт неврологических расстройств и инсульта. 25 мая 2017 г. Проверено 18 марта 2018 г. - ^ «Лейкодистрофия» . www.ninds.nih.gov . Проверено 13 апреля 2024 г.
- ^ Когган, Джей С.; Биттнер, Стефан; Штифель, Клаус М.; Мейт, Свен Г.; Прескотт, Стивен А. (сентябрь 2015 г.). «Физиологическая динамика демиелинизирующих заболеваний: раскрытие сложных взаимосвязей посредством компьютерного моделирования» . Международный журнал молекулярных наук . 16 (9): 21215–21236. дои : 10.3390/ijms160921215 . ISSN 1422-0067 . ПМЦ 4613250 . ПМИД 26370960 .
- ^ Бонковски, Джошуа (24 августа 2010 г.). «Бремя наследственных лейкодистрофий у детей» . Неврология . 75 (8): 718–725. дои : 10.1212/WNL.0b013e3181eee46b . ПМЦ 2931652 . ПМИД 20660364 .
- ^ Jump up to: а б Грациано, AC; Кардил, В. (26 сентября 2014 г.). «История, генетика и последние достижения в области болезни Краббе». Джин . 555 (1): 2–13. дои : 10.1016/j.gene.2014.09.046 . ПМИД 25260228 .
- ^ Роузбуш, ИП (2003). «Поздняя дистония и ее лечение» . Журнал психиатрии и неврологии . 28 (3):240. ПМК 161748 .
- ^ Лю, Ю; Цзоу, Л; Мэн, Ю; Чжан, Ю; Ши, Х; Джу, Дж; Ян, Г; Ху, Л; Чен, X (июнь 2014 г.). «[Семья с двумя детьми с диагнозом аспартилглюкозаминурия - отчет о случае и обзор литературы]». Чжунхуа Эр За Чжи . 52 (6): 455–9. ПМИД 25190167 .
- ^ Турон-Винас, Э; Пинеда, М; Таким образом, В; Лопес-Ласо, Э; Дель Посо, РЛ; Гутьеррес-Солана, LG; Морено, округ Колумбия; Сьерра-Корколес, К; Олабарриета-Ойос, Н.; Мадруга-Гарридо, М; Агирре-Родригес, Дж; Гонсалес-Альварес, В; О'Каллаган, М; Мачарт, Дж; Армстронг-Морон, Дж (13 июля 2014 г.). «Исчезающая болезнь белого вещества у испанской популяции» . J Cent Nerv Syst Dis . 6 : 59–68. дои : 10.4137/JCNSD.S13540 . ПМК 4116383 . ПМИД 25089094 .
- ^ Рубин, Рита (13 марта 2016 г.). «Forbes.com: Масло Лоренцо не могло вылечить Лоренцо, но ожидается, что обследование новорожденных спасет других от его участи» . Форбс.com . Проверено 31 июля 2018 г. .
- ^ Jump up to: а б с д и Биффи, А.; Обур, П.; Картье, Н. (2011). «Генная терапия лейкодистрофий» . Молекулярная генетика человека . 20 (Р1): Р42–Р53. дои : 10.1093/hmg/ddr142 . ПМИД 21459776 .
- ^ Дюшанж, Н.; Дарги, С; д'Одиффре, Д; Кэллис, я; Лапуант, AS; Лоев, Б; Беспфлуг-Танги, О; Мутель, Г. (18 сентября 2014 г.). «Этический менеджмент при создании европейской базы данных по редким заболеваниям лейкодистрофии» . Eur J Paediatr Neurol . 18 (5): 597–603. дои : 10.1016/j.ejpn.2014.04.002 . ПМИД 24786336 . S2CID 31385905 .
- ^ Ян, Эдвард; Прабху, Санджай П. (5 марта 2014 г.). «Визуализирующие проявления лейкодистрофий, наследственных заболеваний белого вещества». Радиологические клиники Северной Америки . 52 (2): 279–319. дои : 10.1016/j.rcl.2013.11.008 . ПМИД 24582341 .
- ^ Jump up to: а б Линь, Шу-Тин; Птачек, Луи Дж.; Фу, Ин-Хуэй (26 января 2011 г.). «Аутосомно-доминантная лейкодистрофия с началом у взрослых: связь ядерной оболочки с миелином» . Журнал неврологии . 31 (4): 1163–1166. doi : 10.1523/jneurosci.5994-10.2011 . ПМК 3078713 . ПМИД 21273400 .
- ^ Коултер-Маки, МБ; Рип, Дж; Лудман, доктор медицинских наук; Бейс, Дж; Коул, Декабрь (октябрь 1995 г.). «Метахроматическая лейкодистрофия (МЛД) у пациента с конституциональной кольцевой хромосомой 22» . Журнал медицинской генетики . 32 (10): 787–91. дои : 10.1136/jmg.32.10.787 . ПМЦ 1051701 . ПМИД 8558556 .
- ^ Сасса, Такаюки; Кихара, Акио (22 марта 2014 г.). «Метаболизм жирных кислот с очень длинной цепью: гены и патофизиология» . Биомолекулы и терапия . 22 (2): 83–92. дои : 10.4062/biomolther.2014.017 . ПМЦ 3975470 . ПМИД 24753812 .
- ^ Jump up to: а б Барбура, Ильхем; Ферчичи, Салима; Дандана, Азза; Джайдан, Зайнеб; Бен Халифа, Сухайра; Чахед, Хинда; Бен Мансур, Рашида; Чебель, Сабля; Мэр Ирен; Милед, Абдельхеди (2010). «Метахроматическая лейкодистрофия. Клинические, биологические и терапевтические аспекты». Анналы клинической биологии . 68 (4): 385–91. дои : 10.1684/abc.2010.0448 . ПМИД 20650733 .
- ^ Гизельман, В; Крагелох-Манн, I (2010). «Метахроматическая лейкодистрофия - обновление». Нейропедиатрия . 41 (1): 1–6. дои : 10.1055/s-0030-1253412 . ПМИД 20571983 .
- ^ Шиманская, Кристина; Луговская, Агнешка; Лора-Камионовска, Милена; Герущак-Бялек, Дорота; Мусиелак, Малгожата; Эйхлер, Сабрина; Гизе, Анн-Катрин; Рольфс, Арндт (2012). «Диагностические трудности при болезни Краббе: отчет о двух случаях и обзор литературы» . Нейропатоловая фольга . 50 (4): 346–356. дои : 10.5114/fn.2012.32364 . ПМИД 23319190 .
- ^ Кольшуттер, Альфрид (25 апреля 2013 г.). «Лизосомальные лейкодистрофии». Детская неврология Часть III . Справочник по клинической неврологии. Том. 113. стр. 1611–1618. дои : 10.1016/B978-0-444-59565-2.00029-0 . ISBN 9780444595652 . ПМИД 23622382 .
- ^ Jump up to: а б Бергер, Дж; Форсс-Петтер, С; Эйхлер, Ф.С. (март 2014 г.). «Патофизиология Х-сцепленной адренолейкодистрофии» . Биохимия . 98 (100): 135–142. дои : 10.1016/j.biochi.2013.11.023 . ПМЦ 3988840 . ПМИД 24316281 .
- ^ Сингх, Навнит; Биксби, Кэтрин; Этьен, Дензил; Таббс, Р. Шейн; Лукас, Мариос (декабрь 2012 г.). «Болезнь Александра: переоценка неонатальной формы». Нервная система ребенка . 28 (12): 2029–2031. дои : 10.1007/s00381-012-1868-8 . ПМИД 22890470 . S2CID 5851209 .
- ^ Хол, Элли М.; Пекны, Милош (февраль 2015 г.). «Глиальный фибриллярный кислый белок (GFAP) и система промежуточных филаментов астроцитов при заболеваниях центральной нервной системы». Современное мнение в области клеточной биологии . 32 (Клеточная архитектура): 121–130. дои : 10.1016/j.ceb.2015.02.004 . ПМИД 25726916 .
- ^ Jump up to: а б Кольшуттер, Альфрид; Эйхлер, Флориан (октябрь 2011 г.). «Детские лейкодистрофии: клиническая перспектива» . Экспертный обзор нейротерапии . 11 (10): 1485–1496. дои : 10.1586/ern.11.135 . ПМИД 21955203 . S2CID 27471268 . [ постоянная мертвая ссылка ]
- ^ Jump up to: а б Пауэлс, PJW; Вандервер, А.; Бернар, Г.; Вольф, Н.; Дреха-Кульчевский, SW; Деони, SCL; Бертини, Э.; Кольшуттер, А.; Ричардсон, В.; френч-Констант, К.; Колер, В.; Баркович, А. (2014). «Гипомиелинирующие лейкодистрофии: прогресс и перспективы трансляционных исследований» (PDF) . Энн. Нейрол . 76 (1): 5–19. дои : 10.1002/ana.24194 . ПМИД 24916848 . S2CID 19026052 .
- ^ «Первый ребенок получает спасительную генную терапию в Национальной системе здравоохранения» . Национальная служба здравоохранения Англии . Национальная служба здравоохранения. 15 февраля 2023 г. Проверено 18 февраля 2023 г.
- ^ Розенберг, Дж.Б.; Каминский С.М.; Обур, П.; Кристалл, РГ; Сондхи, Д. (2016). «Генная терапия метахроматической лейкодистрофии» . Журнал нейробиологических исследований . 94 (11): 1169–79. дои : 10.1002/jnr.23792 . ПМК 5027970 . ПМИД 27638601 .
- ^ Леска, Г; Ванье, Монтана; Крейссон, Э; Бенделак, Н.; Энке, Б; Олланьон-Роман, Э; Обур, П. (август 2005 г.). «Х-сцепленная адренолейкодистрофия у пробанда женского пола: клиническая картина, биологический диагноз и семейные последствия». Архивы педиатрии . 12 (8): 1237–40. дои : 10.1016/j.arcped.2005.03.050 . ПМИД 15878823 .
- ^ «Информационная страница о лейкодистрофии» . www.ninds.nih.gov .
- ^ «Глобальная инициатива по борьбе с лейкодистрофией» . Глобальная инициатива по борьбе с лейкодистрофией .
- ^ «Аккуэйль -» . ela-asso.com .
- ^ "Исследовать" . Объединенный фонд лейкодистрофии .
- ^ «Главная | Метахроматическая лейкодистрофия» . лечение .
- ^ «Фонд МЛД» . mldfoundation.org .
- ^ «leukodystrophyalliance.org — Этот веб-сайт выставлен на продажу! — Ресурсы и информация о leukodystrophyalliance» . leukodystrophyalliance.org .
{{cite web}}: Cite использует общий заголовок ( справка ) - ^ «Пожалуйста, помогите детям с лейкодистрофией» . www.classy.org .
- ^ «Загадочная история братьев Кларк — 4 канал» . Канал 4 . 20 ноября 2012 года. Архивировано из оригинала 20 ноября 2012 года . Проверено 26 ноября 2012 г.
- ^ «Забавный случай с мальчиками, живущими задом наперед» . Независимый . 24 ноября 2012 г. Архивировано из оригинала 14 июня 2022 г. Проверено 2 апреля 2021 г.