Метахроматическая лейкодистрофия
Эта статья нуждается в дополнительных цитатах для проверки . ( январь 2020 г. ) |
| Метахроматическая лейкодистрофия | |
|---|---|
| Другие имена | MLD, дефицит арилсульфатазы А, дефицит ARSA |
 | |
| Сульфатид | |
| Специальность | Эндокринология , неврология |
| Симптомы | Прогрессирующее неврологическое снижение |
| Осложнения | Деменция, судороги, потеря двигательных навыков. |
| Обычное начало | Поздний инфантильный (1-2 года), юношеский (3-20 лет) или взрослый возраст (около 40 лет). |
| Продолжительность | Поздний инфантильный (3-10 лет), юношеский и взрослый (варьируется) |
| Типы | Поздний инфантильный, юношеский или взрослый возраст. |
| Причины | Лизосомальная болезнь накопления |
| Метод диагностики | Ферментная основа и генетика |
| Уход | ТГСК (предсимптоматическая), Генная терапия (поздний инфантильный период), Паллиативная |
| Прогноз | смертельный |
| Частота | 1 из 40 000 рождений |
Метахроматическая лейкодистрофия (МЛД) — лизосомальная болезнь накопления , которую обычно относят к семейству лейкодистрофий , а также к сфинголипидозам, поскольку она влияет на метаболизм сфинголипидов . Лейкодистрофии влияют на рост и/или развитие миелина — жирового покрытия, которое действует как изолятор вокруг нервных волокон в центральной и периферической нервной системе . MLD предполагает накопление сульфата цереброзида . [1] [2] Метахроматическая лейкодистрофия, как и большинство нарушений ферментов, имеет аутосомно-рецессивный тип наследования. [2]
Признаки и симптомы
[ редактировать ]Как и многие другие генетические нарушения, влияющие на липидный обмен, существует несколько форм МЛД: поздняя инфантильная , юношеская и взрослая. [ нужна ссылка ]
- При поздней инфантильной форме , которая является наиболее распространенной формой МЛД (50–60%), у пораженных детей начинаются трудности с ходьбой после первого года жизни, обычно в 15–24 месяца. Симптомы включают мышечную атрофию и слабость, мышечную ригидность , задержку развития, прогрессирующую потерю зрения, приводящую к слепоте, судороги , нарушение глотания, паралич и слабоумие . Дети могут впасть в кому . Без лечения большинство детей с этой формой MLD умирают к 5 годам, часто намного раньше.
- У детей с ювенильной формой МЛД (начало в возрасте от 3 до 10 лет) обычно начинается ухудшение успеваемости в школе, умственное ухудшение и деменция, затем развиваются симптомы, аналогичные поздней инфантильной форме, но с более медленным прогрессированием. Возраст смерти варьируется, но обычно составляет от 10 до 15 лет после появления симптомов. Некоторые пациенты могут жить в течение нескольких десятилетий после начала заболевания. Недавняя тенденция состоит в том, чтобы попытаться различать раннеювенильные (в возрасте 3–7 лет) и поздние ювенильные формы заболевания. Обычно у детей раннего возраста первым симптомом является снижение двигательных навыков, тогда как у детей позднего возраста первыми наблюдаются когнитивные нарушения.
- Взрослая форма обычно начинается после 16 лет, часто на четвертом или пятом десятилетии жизни и проявляется как психическое расстройство или прогрессирующая деменция. МЛД, возникшая у взрослых, обычно прогрессирует медленнее, чем поздние инфантильные и ювенильные формы, с затяжным течением, продолжающимся десять лет и более.
Паллиативная помощь может помочь при многих симптомах и обычно улучшает качество и продолжительность жизни. [ нужна ссылка ]
У носителей низкий уровень ферментов по сравнению с членами их семей («нормальные» уровни варьируются от семьи к семье), но даже низкие уровни ферментов достаточны для переработки сульфатида в организме. [ нужна ссылка ]
Причины
[ редактировать ]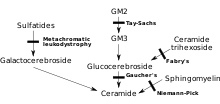
МЛД напрямую вызван дефицитом фермента арилсульфатазы А. [3] (ARSA) и характеризуется активностью ферментов в лейкоцитах менее 10% от нормального контроля. [4] Однако одного только анализа активности фермента ARSA недостаточно для постановки диагноза; Псевдодефицит ARSA, который характеризуется активностью фермента, составляющей 5–20% от нормального контроля, не вызывает MLD. [4] Без этого фермента сульфатиды накапливаются во многих тканях организма, в конечном итоге разрушая миелиновую оболочку нервной системы. Миелиновая оболочка представляет собой жировую оболочку, защищающую нервные волокна. Без него нервы головного мозга (центральная нервная система – ЦНС) и периферические нервы (периферическая нервная система – ПНС), которые контролируют, среди прочего, мышцы, связанные с подвижностью, перестают функционировать должным образом. [ нужна ссылка ]
Арилсульфатаза А активируется сапозином В (Sap B), неферментативным белковым кофактором. [5] Когда уровень фермента арилсульфатазы А нормальный, но сульфатиды все еще высоки (это означает, что они не расщепляются, поскольку фермент не активирован), возникающим заболеванием является дефицит сапозина B, который проявляется аналогично MLD. [4] Дефицит сапозина B встречается очень редко, гораздо реже, чем традиционный MLD. [4] Присутствующий фермент не «способен» работать на нормальном уровне и не может расщеплять сульфатиды, что приводит ко всем тем же симптомам и прогрессированию MLD. [6]
Исследование 2011 года показало, что сульфатид не несет полной ответственности за MLD, поскольку он нетоксичен. Было высказано предположение, что лизосульфатид, сульфатид, у которого удалена ацильная группа, играет определенную роль из-за его цитотоксических свойств in vitro. [7]
Генетика
[ редактировать ]
MLD имеет аутосомно-рецессивный тип наследования. Вероятности наследования в расчете на рождение следующие: [8]
- Если оба родителя являются носителями:
- 25% (1 из 4) детей страдают этим заболеванием.
- 50% (2 из 4) детей будут носителями, но не затронуты заболеванием.
- 25% (1 из 4) детей будут свободны от MLD – здоровый ребенок, не являющийся носителем
- Если один из родителей поражен, а другой свободен от MLD:
- 0% (0) детей будут иметь заболевание – поражен только один родитель, у другого родителя всегда нормальный ген.
- 100% (4 из 4) детей будут носителями (но это не затронет)
- Если один из родителей является носителем, а другой свободен от MLD:
- 50% (2 из 4) детей будут носителями (но это не затронет)
- 50% (2 из 4) детей будут свободны от MLD – здоровый ребенок, не являющийся носителем
Помимо этих частот существует «псевдо»-дефицит, которым страдают 7–15% населения. [9] [10] Люди с псевдодефицитом не имеют никаких проблем с MLD, если только у них не нарушен статус. С помощью текущих диагностических тестов псевдодефицит регистрируется как низкий уровень ферментов, но сульфатид обрабатывается нормально, поэтому симптомов MLD не существует. Это явление наносит ущерб традиционным подходам к скринингу новорожденных , поэтому разрабатываются новые методы скрининга. [ нужна ссылка ]
Диагностика
[ редактировать ]Клиническое обследование и МРТ часто являются первыми шагами в диагностике МЛД. МРТ может указывать на МЛД, но не является адекватным подтверждающим тестом. Анализ крови на уровень фермента ARSA-A с подтверждающим тестом на сульфатид в моче является лучшим биохимическим тестом на МЛД. Подтверждающий сульфатид мочи важен для различения результатов анализа крови MLD и псевдо-MLD. Геномное секвенирование также может подтвердить MLD, однако, вероятно, существует больше мутаций, чем более 200, которые уже известны, вызывающие MLD, которые еще не приписаны MLD, которые вызывают MLD, поэтому в этих случаях биохимический тест все еще оправдан. [ нужна ссылка ]
Скрининг новорожденных
[ редактировать ]Фонд MLD официально запустил инициативу по скринингу новорожденных в конце 2017 года. Разработка скрининга началась в начале 2010-х годов в Вашингтонском университете профессором Майклом Х. Гелбом. Обезличенное пилотное исследование началось в апреле 2016 года в штате Вашингтон. Положительные результаты привели к тому, что MLD был включен в проект ScreenPlus по исследованию детей в штате Нью-Йорк, запуск которого в настоящее время запланирован на первый квартал 2021 года. [ нужна ссылка ]
Уход
[ редактировать ]В настоящее время не существует одобренного лечения MLD у пациентов с клиническими проявлениями в позднем младенческом возрасте или у юношей и взрослых с прогрессирующими симптомами. Существует лечение предсимптомных пациентов и некоторых других пациентов с этим заболеванием.
Пациенты с симптомами обычно получают клиническое лечение, направленное на устранение боли и симптомов. [ нужна ссылка ] Пациенты с предсимптомной поздней инфантильной МЛД, а также пациенты с ювенильной или взрослой МЛД, которые являются либо предсимптомными, либо имеют легкие симптомы, могут рассмотреть возможность трансплантации костного мозга (включая трансплантацию стволовых клеток ), которая может замедлить прогрессирование заболевания в центральном отделе. нервная система. [ нужна ссылка ] Однако результаты в отношении периферической нервной системы были менее драматичными, а долгосрочные результаты этих методов лечения были неоднозначными. [ нужна ссылка ]
В 2020 году Европейское медицинское агентство одобрило препарат клеточной терапии atidarsagene autotemcel (Libmeldy) для лечения инфантильных и ювенильных форм метахроматической лейкодистрофии в Европе. [11] США В 2024 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило атидарсаген аутотемцел (Ленмелди) для лечения предсимптомной поздней инфантильной, предсимптомной ранней ювенильной или ранней симптоматической ювенильной метахроматической лейкодистрофии. [12]
Пресимтомных пациентов можно вылечить с помощью одного курса лечения атидарсагеном аутотемцелом. это разновидность передовой медицины, называемая «генной терапией». Этот тип медицины действует путем доставки генов в организм. Активным веществом атидарсагена аутотемцела являются CD34+ стволовые клетки . Их получают из собственного костного мозга или крови пациента. Затем их модифицируют, чтобы они содержали копию гена, обеспечивающего функциональную ARSA. После подтверждения того, что клетки содержат активную копию гена, их вводят в костный мозг пациента. Клетки CD34+ могут делиться с образованием других типов клеток крови. [11]
Направления исследований
[ редактировать ]В настоящее время изучаются несколько вариантов терапии с использованием клинических испытаний, главным образом, у пациентов позднего детского возраста. Эти методы лечения включают генную терапию , ферментозаместительную терапию (ERT), терапию с уменьшением количества субстрата (SRT) и, возможно, терапию с усилением ферментов (EET). Помимо клинических испытаний, в стадии реализации находится несколько других доклинических исследовательских проектов в области генной терапии. [ нужна ссылка ]
Эпидемиология
[ редактировать ]метахроматической По оценкам , заболеваемость лейкодистрофией встречается у 1 из 40 000–1 из 160 000 человек во всем мире. [13] Заболеваемость гораздо выше в определенных генетически изолированных группах населения, например, 1 из 75 среди хаббанитов (небольшая группа евреев, иммигрировавших в Израиль из южной Аравии), 1 из 2500 в западной части народа навахо и 1 из 8000. среди арабских групп в Израиле. [13]
Поскольку это аутосомно-рецессивное заболевание, 1 из 40 000 соответствует частоте носительства 1 из 100 в общей популяции. [14]
По оценкам, в США ежегодно рождается 3600 детей с MLD, из которых 1900 выживают; в Европе 3100, а во всем мире 49 000 живых. [14]
MLD считается редким заболеванием в США и других странах. [ нужна ссылка ]
Исследовать
[ редактировать ]Терапия трансплантацией костного мозга и стволовых клеток
[ редактировать ]- В настоящее время проводится несколько испытаний, направленных на дальнейшее повышение эффективности и снижение рисков трансплантации костного мозга и стволовых клеток . [ нужна ссылка ]
Генная терапия
[ редактировать ](действительно по состоянию на апрель 2021 г.)
В настоящее время исследуются два разных подхода к генной терапии MLD.
- Генная терапия с трансплантацией аутологичных стволовых клеток . Итальянские исследователи из Института Телемарафона Сан-Раффаэле протестировали новый подход, сочетающий генную терапию с трансплантацией аутологичных стволовых клеток. [15]
- Генная терапия для пациентов позднего младенческого и раннего юношеского возраста одобрена Европейской комиссией [16] в декабре 2020 года после получения положительного заключения Комитета Европейского лекарственного агентства по лекарственным препаратам для использования человеком (CHMP) в октябре 2020 года. [17] [18] Продукт продается в ЕС под названием Libmeldy. Это лекарственный препарат генной терапии, для которого CD34+ гемопоэтические стволовые клетки и клетки-предшественники собираются либо из собственного костного мозга пациента, либо из мобилизованной периферической крови. [19] Эти клетки трансдуцируются ex vivo с использованием лентивирусного вектора, кодирующего ген арилсульфатазы А человека, для вставки функционального гена для продукции фермента ARSA. [19] Когда модифицированные клетки трансплантируются обратно пациенту в виде однократной инфузии, было показано, что клетки производят недостающий фермент ARSA. [19] Дети к пяти годам все были в хорошем состоянии и ходили в детский сад, тогда как обычно к этому возрасту дети с заболеванием даже не могут говорить. [20] Фонда MLD Дополнительную информацию можно найти на странице генной терапии и на сайте Clinical Trials.gov .
- В ноябре 2020 года компания Orchard Therapeutics признала переговоры IND с FDA частью своих усилий по получению одобрения FDA в США. [21]
- Суд над несовершеннолетними был начат в феврале 2020 года. [22]
- Orchard Therapeutics приобрела интеллектуальную собственность на генную терапию у GSK в апреле 2018 года. [23]
- Набор для участия в клинических испытаниях фазы I/II официально начался 24 марта 2010 г. после получения одобрения итальянских властей. Набор первоначальной группы из 8 пациентов был завершен в середине марта 2013 года. Целью исследования была проверка эффективности и безопасности аутологичной (с использованием собственных клеток пациента) трансплантации гемопоэтических стволовых клеток (ТГСК) после генетической модификации для доставки супертерапевтического препарата. сверхэкспрессия) фермента ARSA в нервную систему через клетки крови. Использование собственных стволовых клеток пациента с генетической коррекцией должно уменьшить или устранить осложнения реакции «трансплантат против хозяина» и обеспечить долгосрочное решение правильной экспрессии ARSA у пациентов с MLD. Лабораторные испытания и испытания на животных показали положительные результаты. В июле 2013 года исследователи опубликовали двухлетние результаты первых трех пациентов. Результаты были описаны как многообещающие. [20]
- Клинические испытания фазы I/II завершены. Никакие дополнительные пациенты не набираются, пока данные анализируются и продолжается работа по улучшению технологичности и повторяемости технологии, в то время как рассматривается расширение на другие географические регионы для расширения доступа.
- В апреле 2015 года был завершен набор из 20 пациентов, который включает в себя расширение в декабре 2014 года и добавление еще 6 пациентов.
- Критериями включения являются поздние дети с предсимптомными симптомами, а также подростки как с предсимптомными, так и с ранними симптомами. Подробности о критериях включения и протоколе исследования см. здесь . [24]
- Испытание проходило в одном центре Института Сан-Раффаэле в Милане, Италия. Все расходы должны были оплатить исследователи. Это было трехлетнее исследование. В марте 2013 года последний из 8 пациентов первичного исследования начал терапию. В исследовании приняли участие несколько пациентов с сострадательным доступом, и в конечном итоге оно было расширено до 20 пациентов.
- В конце 2013 года GSK воспользовалась своим правом на использование технологии генной терапии Сан-Рафаэля и работает с миланскими исследователями над подготовкой к следующему этапу исследования. [25]
- Внутримозговая генная терапия – клинические испытания фазы I/II начались в Париже в конце марта 2013 года для участия в клинических испытаниях внутримозговой генной терапии, в ходе которых специальные «векторы», несущие генетически модифицированный материал, вводятся непосредственно в дюжину участков мозга. Есть надежда, что исправленные клетки и фермент, который они производят, затем диффундируют в окружающие области мозга. Обширная лабораторная работа и несколько обнадеживающих исследований ALD легли в основу этого исследования. Впоследствии это судебное разбирательство было прекращено до завершения.
Ферментозаместительная терапия (ФЗТ)
[ редактировать ](действительно по состоянию на февраль 2021 г.)
- Такеда [26] приобрела MLD ERT у Shire в начале 2018 года. [27] и продолжает разрабатывать и изучать свою интратекальную SHP 611 (ранее HGT-1110) ERT (заместительную ферментную терапию).
- Клиническое исследование
- Третье глобальное исследование по изучению поздней инфантильной формы МЛД у 42 пациентов в возрасте от 6 до 72 месяцев началось в апреле 2019 года и было полностью набрано в январе 2021 года. [28] Это первый раз, когда в США открываются учебные центры ERT.
- Фонда MLD Информацию о клинических испытаниях и критерии включения можно найти на странице ERT и на сайте Clinical Trials.gov .
Субстратно-восстановительная терапия
[ редактировать ]- Biomarin South (ранее Zacharon, до того как была приобретена Biomarin в январе 2013 г.) [29] ) из Сан-Диего инициировал программу открытия лекарств для MLD. Эта программа основана на использовании анализов, которые измеряют накопление сульфатидов в культивируемых фибробластах, как средства обнаружения и разработки низкомолекулярных лекарств от MLD. (Этот подход отличается от других подходов, которые измеряют активность ферментов для открытия эффективных лекарств.) По состоянию на июль 2011 года Захарон начал адаптировать методы анализа, разработанные им для других лизосомальных болезней накопления, чтобы их можно было использовать для открытия и разработки лекарств от MLD. (текущий март 2013 г.)
- Система здравоохранения Купера (Нью-Джерси) спонсировала клиническое исследование, проводимое в 2009 году для определения безопасности и эффективности антагониста витамина К (варфарина) при лечении метахроматической лейкодистрофии (МЛД). Никаких результатов опубликовано не было. [30] (текущий март 2013 г.)
Исследования естествознания
[ редактировать ]- В январе 2014 года в Вашингтоне, округ Колумбия, было начато исследование естественной истории болезни (NHS) с участием 30 пациентов, а дополнительные исследовательские центры открылись в США, Европе, Южной Америке, Юго-Восточной Азии и Южной Америке. Из-за проблем с набором участников это исследование было отменено.
- Исследование естествознания проводится в Питтсбурге, штат Пенсильвания, с ноября 2012 года. [31]
Исследования препарата Метазим
[ редактировать ](действительно по состоянию на ноябрь 2023 г.)
Эту статью необходимо обновить . ( ноябрь 2021 г. ) |
Ссылки
[ редактировать ]- ^ «Метахроматическая лейкодистрофия» в Медицинском словаре Дорланда.
- ^ Перейти обратно: а б Ле, Тао; Бхушан, Викас; Хофманн, Джеффри (2012). Первая помощь для USMLE Шаг 1 . МакГроу-Хилл . п. 117 . ISBN 9780071776363 .
- ^ Поппель П., Хабета М., Маркао А., Бюссов Х., Берна Л., Гизельманн В. (март 2005 г.). «Миссенс-мутации как причина метахроматической лейкодистрофии. Деградация арилсульфатазы А в эндоплазматическом ретикулуме» . ФЕБС Дж . 272 (5): 1179–88. дои : 10.1111/j.1742-4658.2005.04553.x . ПМИД 15720392 . S2CID 9371615 .
- ^ Перейти обратно: а б с д Флухарти, Арван. «Дефицит арилсульфатазы А: метахроматическая лейкодистрофия, дефицит ARSA». Джин Ревьюз, 2006 г.
- ^ Кисимото Ю., Хирайва М., О'Брайен Дж.С. (сентябрь 1992 г.). «Сапозины: структура, функции, распределение и молекулярная генетика» . Дж. Липид Рес . 33 (9): 1255–67. дои : 10.1016/S0022-2275(20)40540-1 . ПМИД 1402395 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ «Генетика» . Фонд МЛД. Архивировано из оригинала 22 декабря 2014 г. Проверено 28 мая 2017 г.
- ^ Бломквист, М.; Гизельманн, В.; Монссон, JE (2011). «Накопление лизосульфатида в мозге мышей с дефицитом арилсульфатазы А» . Липиды в здоровье и болезни . 10 (1): 28. дои : 10.1186/1476-511X-10-28 . ПМК 3041674 . ПМИД 21299873 .
- ^ «Аутосомно-рецессивный тип: Медицинская энциклопедия MedlinePlus» . medlineplus.gov . Проверено 18 августа 2021 г.
- ^ Хоэншуц, К; Эйх П; Фридл В; Вахид А; Конзельманн Э; Проппинг П. (апрель 1989 г.). «Псевдодефицит арилсульфатазы А». Генетика человека . 82 (1): 45–8. дои : 10.1007/bf00288270 . ПМИД 2565866 . S2CID 32274162 .
- ^ Герц, Барбара; Бах, Г. (1984). «Арилсульфатаза А при псевдодефиците». Генетика человека . 66 (2–3): 147–150. дои : 10.1007/BF00286589 . ПМИД 6143719 . S2CID 2349721 .
- ^ Перейти обратно: а б «Libmeldy EPAR» , ema.europa.eu , 13 октября 2020 г.
- ^ «FDA одобрило первую генную терапию для детей с метахроматической лейкодистрофией» (пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 18 марта 2024 г. Проверено 20 марта 2024 г.
- ^ Перейти обратно: а б Метахроматическая лейкодистрофия в Домашнем справочнике генетики. Проверено в сентябре 2007 г.
- ^ Перейти обратно: а б «MLD 101: Генетика» . www.mldfoundation.org . 6 января 2017. Архивировано из оригинала 30 декабря 2013 года . Проверено 6 января 2017 г.
- ^ Биффи А., Луккини Г., Ровелли А., Сесса М. (октябрь 2008 г.). «Метахроматическая лейкодистрофия: обзор текущих и перспективных методов лечения» . Трансплантация костного мозга . 42 (Приложение 2): С2–6. дои : 10.1038/bmt.2008.275 . ПМИД 18978739 .
- ^ «Orchard Therapeutics получила одобрение ЕС на препарат Libmeldy для лечения метахроматической лейкодистрофии с ранним началом (MLD)» (пресс-релиз). Садовая терапия. 21 декабря 2020 г. Проверено 12 января 2021 г. - через GlobeNewswire.
- ^ Фруктовый сад, Терапия. «Orchard Therapeutics получила положительное заключение CHMP на препарат Libmeldy для лечения метахроматической лейкодистрофии с ранним началом (MLD)» . № 16 октября 2020 г. Проверено 12 января 2021 г.
- ^ Американский фармацевтический обзор. «Orchard Therapeutics объявляет о регистрации в MAA препарата для лечения метахроматической лейкодистрофии» . Американский фармацевтический обзор . Сравнить сети . Проверено 3 декабря 2019 г.
- ^ Перейти обратно: а б с «Новая генная терапия для лечения редкого генетического заболевания — метахроматической лейкодистрофии» . Европейское агентство лекарственных средств (EMA) (пресс-релиз). 16 октября 2020 г. Проверено 16 октября 2020 г. Текст скопирован из источника, права на который принадлежат Европейскому агентству по лекарственным средствам. Воспроизведение разрешено при условии указания источника.
- ^ Перейти обратно: а б Биффи А., Монтини Е., Лориоли Л. и др. (2013). «Лентивирусная генная терапия гемопоэтическими стволовыми клетками приносит пользу при метахроматической лейкодистрофии» . Наука . 341 (6148): 1233158. doi : 10.1126/science.1233158 . ПМИД 23845948 . S2CID 206546808 .
- ^ «Orchard Therapeutics объявляет о разрешении FDA заявки IND на OTL-200 для лечения метахроматической лейкодистрофии (MLD)» (пресс-релиз). Садовая терапия. 19 ноября 2020 г. Проверено 12 января 2021 г. - через GlobeNewswire.
- ^ «OTL-200 у пациентов с поздней ювенильной метахроматической лейкодистрофией (MLD)» . ClinicalTrials.Gov . Проверено 25 февраля 2020 г.
- ^ Орчард, Терапия (12 апреля 2018 г.). «GSK подписывает стратегическое соглашение о передаче портфеля генной терапии редких заболеваний компании Orchard Therapeutics» . Проверено 12 января 2021 г.
- ^ «Генная терапия MLD - Сан-Раффаэле - Фонд MLD» . Фонд МЛД .
- ^ «Продуктовый конвейер ГСК» . ГСК . Март 2014 года . Проверено 29 июня 2014 г.
- ^ «Трубопровод Такеда)» . Трубопровод Такеда . Проверено 12 сентября 2020 г.
- ^ «Takeda завершает приобретение компании Shire, становясь глобальным, основанным на ценностях и ориентированным на исследования и разработки биофармацевтическим лидером» . Такеда.com . Проверено 7 января 2018 г.
- ^ «Исследование интратекального SHP611 у участников с поздней детской метахроматической лейкодистрофией (Эмболден)» . ClinicalTrails.gov . Проверено 30 апреля 2019 г.
- ^ «BioMarin приобретает Zacharon Pharmaceuticals (NASDAQ:BMRN)» . Архивировано из оригинала 29 января 2013 г. Проверено 16 марта 2013 г.
- ^ «Эффект варфарина в лечении метахроматической лейкодистрофии - полнотекстовый просмотр - ClinicalTrials.gov» . www.clinicaltrials.gov . 18 марта 2011 г.
- ^ «NDRD: Программа изучения развития нервной системы при редких заболеваниях» . NDRD: Программа изучения развития нервной системы при редких заболеваниях . Архивировано из оригинала 1 октября 2020 года . Проверено 12 сентября 2020 г.
- ^ «Открытое расширенное исследование рекомбинантной человеческой арилсульфатазы А (HGT-1111) при позднем детском MLD» . ClinicalTrials.gov. 21 мая 2008 года . Проверено 3 ноября 2021 г.
- ^ «Многоцентровое исследование HGT-1110, вводимого интратекально детям с метахроматической лейкодистрофией (MLD) (IDEAMLD)» . ClinicalTrials.gov. 13 января 2012 года . Проверено 3 ноября 2021 г.
- ^ «Родители больного ребенка потратили 1,75 миллиона долларов на фармацевтическую фирму» . Сидней Морнинг Геральд . 16 июля 2009 года . Проверено 3 ноября 2021 г.
- ^ Джонкхир, Ан И.; Кингма, Сандра Д.К.; Эйскенс, Франсуа; Бордон, Виктория; Янсен, Анна К. (01 сентября 2023 г.). «Метахроматическая лейкодистрофия: проверять или не проверять?» . Европейский журнал детской неврологии . 46 : 1–7. дои : 10.1016/j.ejpn.2023.06.005 . ISSN 1090-3798 .
- Некоторые части этой статьи любезно предоставлены общедоступным текстом, доступным в Национальном институте неврологических расстройств и инсульта :
- «Информационная страница NINDS по метахроматической лейкодистрофии» . Архивировано из оригинала 3 июня 2009 г. Проверено 7 июня 2009 г.