Патофизиология болезни Паркинсона
| Гибель нейронов при БП головного мозга | |
|---|---|
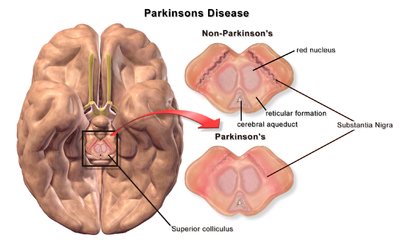 | |
| Мозг без болезни Паркинсона и с ней в сравнении в черной субстанции |
Патофизиология болезни Паркинсона заключается в гибели дофаминергических в нейронов результате изменения биологической активности головного мозга по отношению к болезни Паркинсона (БП). Существует несколько предполагаемых механизмов нейронов гибели при БП; однако не все из них хорошо поняты. Пять предполагаемых основных механизмов гибели нейронов при болезни Паркинсона включают агрегацию белков в тельцах Леви , нарушение аутофагии , изменения клеточного метаболизма или функции митохондрий , нейровоспаление и разрушение гематоэнцефалического барьера (ГЭБ), приводящее к неплотности сосудов. [ 1 ]
Агрегация белков
[ редактировать ]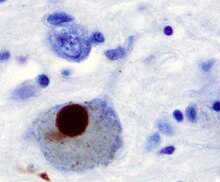
Первой основной предполагаемой причиной гибели нейронов при болезни Паркинсона является объединение или олигомеризация белков . Белок альфа-синуклеин увеличился в мозге пациентов с болезнью Паркинсона и, поскольку альфа-синуклеин нерастворим, он агрегирует с образованием телец Леви (показано слева) в нейронах. Традиционно считалось, что тельца Леви являются основной причиной гибели клеток при болезни Паркинсона; однако более поздние исследования показывают, что тельца Леви приводят к другим эффектам, вызывающим гибель клеток. [ 2 ] Тем не менее, тельца Леви широко признаны патологическим маркером болезни Паркинсона.
Тельца Леви сначала появляются в обонятельной луковице , продолговатом мозге и покрышке моста ; пациенты на этой стадии протекают бессимптомно. По мере прогрессирования заболевания в черной субстанции , областях среднего и переднего мозга , а также в неокортексе развиваются тельца Леви .
Этот механизм подтверждается тем, что α-синуклеин нетоксичен, когда не способен образовывать агрегаты; что белки теплового шока, которые помогают в рефолдинге белков, чувствительных к агрегации, благотворно влияют на ПД при сверхэкспрессии; и что реагенты, которые нейтрализуют агрегированные виды, защищают нейроны в клеточных моделях сверхэкспрессии α-синуклеина. [ 3 ]
Альфа-синуклеин , по-видимому, является ключевым связующим звеном между снижением репарации ДНК и болезнью Паркинсона. [ 4 ] Альфа-синуклеин активирует ATM ( мутация атаксии-телеангиэктазии ), основную повреждений ДНК восстановления сигнальную киназу . Альфа-синуклеин связывается с разрывами двухцепочечной ДНК и облегчает процесс репарации ДНК при соединении негомологичных концов . [ 5 ] Было предложено [ 5 ] что цитоплазматическая агрегация альфа-синуклеина с образованием телец Леви что приводит к снижению репарации ДНК, увеличению двухцепочечных разрывов ДНК и увеличению запрограммированной гибели клеток нейронов снижает его ядерные уровни , .
Нарушение аутофагии
[ редактировать ]
Второй основной предполагаемый механизм гибели нейронов при болезни Паркинсона — аутофагия — представляет собой механизм, посредством которого внутренние компоненты клетки расщепляются и перерабатываются для использования. [ 2 ] [ 6 ] Было доказано, что аутофагия играет важную роль в здоровье мозга, помогая регулировать клеточную функцию. Нарушение механизма аутофагии может привести к нескольким различным типам заболеваний, таких как болезнь Паркинсона. [ 6 ] [ 7 ]
Также было показано, что дисфункция аутофагии при болезни Паркинсона приводит к нарушению регуляции деградации митохондрий . [ 8 ]
Изменения клеточного метаболизма
[ редактировать ]
Третья основная предполагаемая причина гибели клеток при болезни Паркинсона связана с генерирующей энергию органеллой митохондрии . При болезни Паркинсона функция митохондрий нарушается, что подавляет выработку энергии и приводит к смерти. [ 9 ] [ 10 ]
Предполагается, что механизм митохондриальной дисфункции при болезни Паркинсона сосредоточен в PINK1 и комплексе Паркина , поскольку было показано, что это управляет аутофагией митохондрий (также известной как митофагия ). [ 9 ] [ 10 ] [ 11 ] PINK1 — это белок, который обычно транспортируется в митохондрии, но может также накапливаться на поверхности поврежденных митохондрий. Накопленный PINK1 затем рекрутирует Паркина; Паркин инициирует разрушение дисфункциональных митохондрий — механизм, который действует как «контроль качества». [ 9 ] Считается, что при болезни Паркинсона гены, кодирующие PINK1 и Parkin, мутируют, что ухудшает способность этих белков разрушать дисфункциональные митохондрии, что приводит к нарушению функции и морфологии митохондрий и, в конечном итоге, к гибели клеток. [ 9 ] [ 10 ] Также было показано, что мутации митохондриальной ДНК (мтДНК) накапливаются с возрастом. [ 12 ] что указывает на то, что восприимчивость к этому механизму гибели нейронов увеличивается с возрастом.
Еще одним механизмом гибели клеток при болезни Паркинсона, связанным с митохондриями, является образование активных форм кислорода (АФК). [ 12 ] [ 13 ] АФК — это высокореактивные молекулы, которые содержат кислород и могут нарушать функции митохондрий и остальной части клетки. С возрастом митохондрии теряют способность удалять АФК, но продолжают производить АФК, вызывая увеличение чистого производства АФК и, в конечном итоге, гибель клеток. [ 12 ] [ 13 ]
Согласно обзору Puspita et al. [ 14 ] исследования показали, что в и эндоплазматическом ретикулуме митохондриях уровни альфа-синуклеина и дофамина , вероятно, участвуют в развитии окислительного стресса , а также симптомов БП. Окислительный стресс , по-видимому, играет роль в опосредовании отдельных патологических событий, которые вместе в конечном итоге приводят к гибели клеток при БП. [ 14 ] Окислительный стресс, приводящий к гибели клеток, может быть общим знаменателем, лежащим в основе многих процессов. Окислительный стресс вызывает окислительное повреждение ДНК . Такое повреждение увеличивается в митохондриях черной субстанции пациентов с БП и может привести к гибели клеток черных нейронов. [ 15 ] [ 16 ]
Нейровоспаление
[ редактировать ]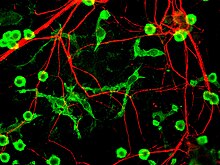
Четвертый предполагаемый основной механизм гибели нейронов при болезни Паркинсона, нейровоспаление , в целом понятен для нейродегенеративных заболеваний, однако конкретные механизмы болезни Паркинсона не полностью охарактеризованы. [ 17 ] Одним из основных типов клеток, участвующих в нейровоспалении, является микроглия . Микроглия признана врожденными иммунными клетками центральной нервной системы . Микроглия активно исследует окружающую среду и значительно меняет морфологию своих клеток в ответ на повреждение нейронов. Острое воспаление в головном мозге обычно характеризуется быстрой активацией микроглии. В этот период периферический иммунный ответ отсутствует. Однако со временем хроническое воспаление вызывает деградацию тканей и гематоэнцефалического барьера. В это время микроглия генерирует активные формы кислорода и высвобождает сигналы для привлечения периферических иммунных клеток для воспалительной реакции.
Кроме того, известно, что микроглия имеет два основных состояния: M1 — состояние, при котором клетки активируются и выделяют провоспалительные факторы; и М2 — состояние, при котором клетки деактивируются и выделяют противовоспалительные факторы. [ 18 ] Микроглия обычно находится в состоянии покоя (М2), но при болезни Паркинсона может переходить в М1 благодаря наличию агрегатов α-синуклеина. Микроглия M1 высвобождает провоспалительные факторы, которые могут привести к гибели мотонейронов. В этом случае умирающие клетки могут высвободить факторы, увеличивающие активацию микроглии M1, что приводит к возникновению петли положительной обратной связи , которая вызывает постоянно увеличивающуюся гибель клеток. [ 17 ]
пробой ГЭБ
[ редактировать ]
Пятый предполагаемый основной механизм гибели клеток — это разрушение гематоэнцефалического барьера (ГЭБ). ГЭБ имеет три типа клеток, которые жестко регулируют поток молекул в мозг и из него: эндотелиальные клетки , перициты и астроциты . При нейродегенеративных заболеваниях распад ГЭБ измеряли и идентифицировали в определенных областях мозга, включая черную субстанцию при болезни Паркинсона и гиппокамп при болезни Альцгеймера. [ 19 ] Белковые агрегаты или цитокины нейровоспаления могут влиять на клеточные рецепторы и изменять их функцию в ГЭБ. [ 19 ] [ 20 ] В частности, фактора роста эндотелия сосудов (VEGF) и рецепторов VEGF считается, что при нейродегенеративных заболеваниях нарушается регуляция . Взаимодействие между белком VEGF и его рецепторами приводит к пролиферации клеток, но считается, что оно нарушается при болезни Паркинсона и болезни Альцгеймера. [ 20 ] [ 21 ] Затем это приводит к остановке роста клеток и, следовательно, к предотвращению образования новых капилляров посредством ангиогенеза . Нарушение клеточных рецепторов также может повлиять на способность клеток прикрепляться друг к другу с помощью слипчивых соединений . [ 22 ]
Без образования новых капилляров существующие капилляры разрушаются, и клетки начинают диссоциировать друг от друга. Это, в свою очередь, приводит к разрушению щелевых соединений. [ 23 ] [ 24 ] Щелевые соединения в эндотелиальных клетках ГЭБ помогают предотвратить попадание крупных или вредных молекул в мозг, регулируя поток питательных веществ в мозг. Однако по мере разрушения щелевых контактов белки плазмы могут проникать во внеклеточный матрикс мозга. [ 23 ] Этот механизм также известен как непроницаемость сосудов, при которой дегенерация капилляров приводит к «протеканию» крови и белков крови в мозг. Неплотность сосудов может в конечном итоге привести к изменению функции нейронов и переходу в сторону апоптотического поведения или гибели клеток.
Влияние на передвижение
[ редактировать ]
Дофаминергические нейроны — наиболее распространенный тип нейронов черной субстанции , части мозга, регулирующей двигательный контроль и обучение. Дофамин — нейромедиатор , который модулирует активность мотонейронов центральной нервной системы . Активированные мотонейроны затем передают свои сигналы через потенциал действия мотонейронам спинного мозга. [ 25 ] Однако когда погибает значительный процент мотонейронов (около 50-60%), уровень дофамина снижается до 80%. [ 10 ] Это подавляет способность нейронов генерировать и передавать сигнал. Это подавление передачи в конечном итоге вызывает характерную паркинсоническую походку с такими симптомами, как сгорбленная и замедленная ходьба или тремор.
Ссылки
[ редактировать ]- ^ Тэнси М.Г., Голдберг М.С. (2010). «Нейровоспаление при болезни Паркинсона: его роль в гибели нейронов и последствия для терапевтического вмешательства» . Нейробиология болезней . 37 (3): 510–518. дои : 10.1016/j.nbd.2009.11.004 . ПМЦ 2823829 . ПМИД 19913097 .
- ^ Перейти обратно: а б Шапира А.Х. (2009). «Этиология и патогенез болезни Паркинсона». Неврологические клиники . 27 (3): 583–603. дои : 10.1016/j.ncl.2009.04.004 .
- ^ Стефанис, Леонидас (2012). «А-Синуклеин при болезни Паркинсона» . Колд Спринг Харб Перспектив Мед . 4 (2): а009399. doi : 10.1101/cshperspect.a009399 . ПМЦ 3281589 . ПМИД 22355802 .
- ^ Abugable AA, Моррис Дж.Л., Пальминья Н.М., Заксаускайте Р., Рэй С., Эль-Хамиси С.Ф. (сентябрь 2019 г.). «Репарация ДНК и неврологические заболевания: от молекулярного понимания к разработке диагностики и модельных организмов» . Восстановление ДНК (Амст.) . 81 : 102669. doi : 10.1016/j.dnarep.2019.102669 . ПМИД 31331820 .
- ^ Перейти обратно: а б Шазер А.Дж., Остерберг В.Р., Дент С.Е., Стэкхаус Т.Л., Уэйкхэм СМ, Бутрос С.В., Уэстон Л.Дж., Оуэн Н., Вайсман Т.А., Луна Е, Рабер Дж., Люк К.С., Маккалоу А.К., Вольтьер Р.Л., Унни В.К. (июль 2019 г.). «Альфа-синуклеин — это ДНК-связывающий белок, который модулирует восстановление ДНК, что приводит к нарушениям с тельцами Леви» . наук. Представитель . 9 (1): 10919. doi : 10.1038/s41598-019-47227-z . ПМК 6662836 . ПМИД 31358782 .
- ^ Перейти обратно: а б Стерн С.Т., Джонсон Д.Н. (2008). «Роль взаимодействия наноматериала и аутофагии при нейродегенеративных заболеваниях» . Аутофагия . 4 (8): 1097–1100. дои : 10.4161/авто.7142 .
- ^ Гавами С., Шоджаи С., Йегане Б., Анде С.Р., Джангамредди Дж.Р., Мехпур М., Лос М.Дж. (2014). «Аутофагия и дисфункция апоптоза при нейродегенеративных заболеваниях» . Прогресс нейробиологии . 112 : 24–49. дои : 10.1016/j.pneurobio.2013.10.004 .
- ^ Ху Цзы, Ян Б, Мо Х, Сяо Х (2014). «Механизм и регуляция аутофагии и ее роль при заболеваниях нейронов». Молекулярная нейробиология . 52 (3): 1190–1209. дои : 10.1007/s12035-014-8921-4 .
- ^ Перейти обратно: а б с д Чен Х, Чан, округ Колумбия (2009). «Митохондриальная динамика - слияние, деление, движение и митофагия - при нейродегенеративных заболеваниях» . Молекулярная генетика человека . 18 (С2): Р169–Р176. дои : 10.1093/hmg/ddp326 . ПМЦ 2758711 . ПМИД 19808793 .
- ^ Перейти обратно: а б с д Пикрелл А., Юл Р. (2015). «Роль PINK1, Паркина и митохондриальной точности при болезни Паркинсона» . Нейрон . 85 (2): 257–273. дои : 10.1016/j.neuron.2014.12.007 . ПМЦ 4764997 . ПМИД 25611507 .
- ^ Нарендра Д., Танака А., Суен Д., Юл Р.Дж. (2008). «Паркин избирательно рекрутируется в поврежденные митохондрии и способствует их аутофагии» . Журнал клеточной биологии . 183 (5): 795–803. дои : 10.1083/jcb.200809125 . ПМК 2592826 . ПМИД 19029340 .
- ^ Перейти обратно: а б с Лин М.Т., Бил М.Ф. (2006). «Митохондриальная дисфункция и окислительный стресс при нейродегенеративных заболеваниях». Природа . 443 (7113): 787–795. дои : 10.1038/nature05292 . ПМИД 17051205 .
- ^ Перейти обратно: а б Джомова К., Вондракова Д., Лоусон М., Валко М. (2010). «Металлы, окислительный стресс и нейродегенеративные расстройства». Мол Клеточная Биохимия . 345 (1–2): 91–104. дои : 10.1007/s11010-010-0563-x .
- ^ Перейти обратно: а б Пуспита Л., Чунг С.Ю., Шим Дж.В. (ноябрь 2017 г.). «Окислительный стресс и клеточные патологии при болезни Паркинсона» . Мол Брейн . 10 (1): 53. дои : 10.1186/s13041-017-0340-9 . ПМК 5706368 . ПМИД 29183391 .
- ^ Шимура-Миура Х., Хаттори Н., Канг Д., Мияко К., Накабеппу Ю., Мизуно Ю. (декабрь 1999 г.). «Повышение уровня 8-оксо-дГТФазы в митохондриях нейронов черной субстанции при болезни Паркинсона». Энн. Нейрол . 46 (6): 920–4. ПМИД 10589547 .
- ^ Накабеппу Ю, Цучимото Д, Ямагути Х, Сакуми К (апрель 2007 г.). «Окислительное повреждение нуклеиновых кислот и болезнь Паркинсона». Дж. Нейроски. Рез 85 (5): 919–34. дои : 10.1002/jnr.21191 . hdl : 2324/8296 . ПМИД 17279544 .
- ^ Перейти обратно: а б Гласс СК, Сайджо К, Победитель Б, Маркетто МС, Гейдж ФХ (2010). «Механизмы, лежащие в основе воспаления при нейродегенерации» . Клетка . 140 (6): 918–934. дои : 10.1016/j.cell.2010.02.016 . ПМЦ 2873093 . ПМИД 20303880 .
- ^ Злокович Б.В. (2008). «Гематоэнцефалический барьер в здоровье и хронических нейродегенеративных заболеваниях» . Нейрон . 57 (2): 178–201. дои : 10.1016/j.neuron.2008.01.003 .
- ^ Перейти обратно: а б Злокович Б.В. (2011). «Нейроваскулярные пути нейродегенерации при болезни Альцгеймера и других расстройствах» . Nat Rev Neurosci . 12 (12): 723–738. дои : 10.1038/nrn3114 . ПМК 4036520 . ПМИД 22048062 .
- ^ Перейти обратно: а б Хао Т., Роквелл П. (2013). «Передача сигналов через рецептор фактора роста эндотелия сосудов VEGFR-2 защищает нейроны гиппокампа от митохондриальной дисфункции и окислительного стресса» . Свободнорадикальная биология и медицина . 63 : 421–431. doi : 10.1016/j.freeradbiomed.2013.05.036 . ПМЦ 3756493 . ПМИД 23732519 .
- ^ Альмодовар Ч.Р., Ламбрехтс Д., Маццоне М., Кармелиет П. (2009). «Роль и терапевтический потенциал VEGF в нервной системе». Физиологические обзоры . 89 (2): 607–648. doi : 10.1152/physrev.00031.2008 .
- ^ Фёрстер С., Бурек М., Ромеро И.А., Векслер Б., Куро П., Дренкхан Д. (2008). «Дифференциальное воздействие гидрокортизона и TNFα на белки плотных соединений в модели гематоэнцефалического барьера человека in vitro» . Журнал физиологии . 586 (7): 1937–1949. дои : 10.1113/jphysicalol.2007.146852 . ПМЦ 2375735 . ПМИД 18258663 .
- ^ Перейти обратно: а б Нагасава К., Тиба Х., Фудзита Х., Кодзима Т., Сайто Т., Эндо Т., Савада Н. (2006). «Возможное участие щелевых соединений в барьерной функции плотных соединений эндотелиальных клеток головного мозга и легких». Журнал клеточной физиологии . 208 (1): 123–132. дои : 10.1002/jcp.20647 .
- ^ Марамбо П., Дрезес-Верринглоер У., Винтде В. (2009). «Передача сигналов кальция при нейродегенерации» . Мол Нейродегенер . 4 (1): 20. дои : 10.1186/1750-1326-4-20 . ПМЦ 2689218 . ПМИД 19419557 .
- ^ Барнетт М.В., Ларкман П.М., Ларкман (2007). «Потенциал действия». Практика Нейрол . 7 (3): 192–7. ПМИД 17515599 .