синдром Гурлера
| синдром Гурлера | |
|---|---|
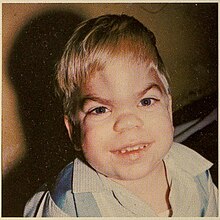 | |
| Пациент с синдромом Гурлера | |
| Причины | Дефицит фермента альфа-L идуронидазы. |
| Дифференциальный диагноз | синдром Гурлера-Шейе ; синдром Шейе ; синдром Хантера ; другие мукополисахаридозы |
| Прогноз | Смерть обычно наступает до 12 лет. |
| Частота | 1 из 100 000 |
Синдром Гурлера , также известный как мукополисахаридоз типа IH ( MPS-IH ), болезнь Гурлера и ранее гаргоилизм , представляет собой генетическое заболевание , которое приводит к накоплению больших молекул сахара, называемых гликозаминогликанами (ГАГ) в лизосомах . Неспособность расщеплять эти молекулы приводит к широкому спектру симптомов, вызванных повреждением нескольких различных систем органов , включая, помимо прочего, нервную систему , скелетную систему , глаза и сердце .
Основной механизм — дефицит альфа-L-идуронидазы , фермента, ответственного за расщепление ГАГ. [1] : 544 накопление дерматансульфата и гепарансульфата Без этого фермента в организме происходит . Симптомы появляются в детстве, и обычно наступает ранняя смерть. Другие, менее тяжелые формы МПС I типа включают синдром Гурлера-Шейе (MPS-IHS) и синдром Шейе (MPS-IS).
Синдром Гурлера классифицируется как лизосомальная болезнь накопления . Клинически это связано с синдромом Хантера (МПС II); [2] однако синдром Хантера является Х-сцепленным , тогда как синдром Гурлера является аутосомно-рецессивным .
Признаки и симптомы
[ редактировать ]
Дети с синдромом Гурлера могут выглядеть нормальными при рождении, а симптомы у него развиваются в первые годы жизни. Симптомы различаются у разных пациентов. [ нужна ссылка ]
Одним из первых обнаруженных отклонений является огрубление черт лица; эти симптомы могут начаться в возрасте 3–6 месяцев. Голова может быть большой с выступающими лобными костями . Череп может быть удлиненным . Нос может иметь уплощенную переносицу с постоянными выделениями из носа. Глазницы могут быть широко расставлены, а глаза могут выступать из черепа. Губы могут быть большими, и больные дети могут постоянно держать челюсти открытыми. Скелетные аномалии возникают примерно в возрасте 6 месяцев, но могут не проявляться клинически до 10–14 месяцев. Пациенты могут испытывать изнурительные деформации позвоночника и бедра, туннельный синдром запястья и тугоподвижность суставов. Пациенты могут иметь нормальный рост в младенчестве, но перестают расти к двум годам. Они не могут достигать высоты более 4 футов (1,2 м). [ нужна ссылка ]
Другие ранние симптомы могут включать паховые и пупочные грыжи . Они могут присутствовать при рождении или развиться в течение первых месяцев жизни. Помутнение роговицы и дегенерация сетчатки могут возникнуть в течение первого года жизни, что приводит к слепоте. увеличение печени и селезенки Часто наблюдается . Дисфункции органов нет, но отложение ГАГ в этих органах может привести к значительному увеличению их размеров. У пациентов также может наблюдаться диарея . аортального клапана . Может возникнуть заболевание [ нужна ссылка ]
Часто возникает обструкция дыхательных путей, обычно вторичная по отношению к аномалиям шейных позвонков. [3] Часто могут возникать инфекции верхних и нижних дыхательных путей. [ нужна ссылка ]
Задержка развития может стать очевидной в возрасте 1–2 лет, при этом максимальный функциональный возраст составляет 2–4 года. Далее следует прогрессивное ухудшение. У большинства детей развиваются ограниченные речевые способности. Смерть обычно наступает к 10 годам. [4] [5]
Генетика
[ редактировать ]
Дети с синдромом Гурлера несут две дефектные копии гена IDUA , который картирован в участке 4p16.3 на хромосоме 4 . Это ген, который кодирует белок идуронидазу. По состоянию на 2018 год [update] , что более 201 различных мутаций в гене IDUA вызывают МПС I. Было показано [6]
Поскольку синдром Гурлера является аутосомно- рецессивным заболеванием, у больных имеются две нерабочие копии гена. Человек, рожденный с одной нормальной копией и одной дефектной копией, называется носителем . Они будут производить меньше α-L-идуронидазы, чем человек с двумя нормальными копиями гена. Однако сниженное производство фермента у носителей остается достаточным для нормального функционирования; у человека не должно быть никаких симптомов заболевания. [ нужна ссылка ]
Механизмы
[ редактировать ]
Ген IDUA отвечает за кодирование фермента альфа-L-идуронидазы. Посредством гидролиза альфа-L-идуронидаза отвечает за расщепление молекулы, называемой несульфатированной альфа-L-идуроновой кислотой . Это уроновая кислота , содержащаяся в ГАГ, дерматансульфате и гепарансульфате. Фермент альфа-L-идуронидаза локализован в лизосомах. Без достаточной ферментативной функции эти ГАГ не могут быть переварены должным образом. [7]
Диагностика
[ редактировать ]Диагноз часто можно поставить посредством клинического осмотра и анализа мочи (избыток мукополисахаридов выводится с мочой ). Ферментные анализы (тестирование различных клеток или жидкостей организма в культуре на дефицит ферментов) также используются для окончательного диагноза одного из мукополисахаридозов. Пренатальная диагностика с использованием амниоцентеза и отбора проб ворсин хориона позволяет проверить, несет ли плод копию дефектного гена или страдает этим заболеванием. Генетическое консультирование может помочь родителям, у которых в семейном анамнезе есть мукополисахаридозы, определить, являются ли они носителями мутированного гена, вызывающего заболевание. [ нужна ссылка ]
Классификация
[ редактировать ]Все представители семейства мукополисахаридозов также являются лизосомальными болезнями накопления . Мукополисахаридоз I типа (МПС I) подразделяют на три подтипа в зависимости от тяжести симптомов. Все три типа приводят к отсутствию или снижению функционирования одного и того же фермента. МПС-ИГ (синдром Гурлера) — наиболее тяжелый из подтипов МПС I. Два других типа — это MPS-IS ( синдром Шейе ) и MPS-IHS ( синдром Гурлера-Шейе ). [ нужна ссылка ]
Из-за существенного совпадения между синдромом Гурлера, синдромом Гурлера-Шейе и синдромом Шейе некоторые источники считают эти термины устаревшими. Вместо этого МПС I можно разделить на «тяжелую» и «ослабленную» формы. [8]
Уход
[ редактировать ]В настоящее время не существует лечения синдрома Гурлера. Заместительная ферментная терапия идуронидазой (альдуразимом) может улучшить функцию легких и их подвижность. Это может уменьшить количество углеводов, неправильно хранящихся в органах. Может потребоваться хирургическая коррекция деформаций кистей и стоп. Операция на роговице может помочь облегчить проблемы со зрением. [5]
Трансплантация костного мозга (ТКМ) и трансплантация пуповинной крови (UCBT) могут использоваться в качестве лечения МПС I. ТКМ от братьев и сестер с идентичными генами HLA и от родственников со схожими генами HLA может значительно улучшить выживаемость, когнитивные функции и физические симптомы. У пациентов может развиться реакция «трансплантат против хозяина» ; это более вероятно у доноров, не являющихся братьями и сестрами. В исследовании 1998 года у детей с HLA-идентичными донорами-братьями и сестрами пятилетняя выживаемость составила 75%; дети с донорами, не являющимися братьями и сестрами, имели пятилетнюю выживаемость 53%. [9]
Детям часто не хватает доступа к подходящему донору костного мозга. В этих случаях UCBT от неродственных доноров может увеличить выживаемость, уменьшить физические признаки заболевания и улучшить когнитивные функции. Осложнения от этого лечения могут включать реакцию «трансплантат против хозяина» . [10]
Прогноз
[ редактировать ]Британское исследование 2008 года показало, что средняя ожидаемая продолжительность жизни пациентов с синдромом Гурлера составляет 8,7 года. Для сравнения, средняя продолжительность жизни для всех форм МПС I типа составила 11,6 года. Пациенты, которым была проведена успешная трансплантация костного мозга, имели двухлетнюю выживаемость 68% и 10-летнюю выживаемость 64%. У пациентов, которым не была проведена трансплантация костного мозга, продолжительность жизни значительно сократилась: средний возраст составил 6,8 года. [4]
Эпидемиология
[ редактировать ]Общая частота синдрома Гурлера составляет один случай на 100 000 человек. [5] В совокупности все мукополисахаридозы встречаются примерно в одном случае на каждые 25 000 рождений в Соединенных Штатах. [2]
Исследовать
[ редактировать ]Генная терапия
[ редактировать ]Существует большой интерес к лечению МПС I с помощью генной терапии . На животных моделях доставка гена идуронидазы осуществлялась с помощью ретровирусных , аденовирусных , аденоассоциированных вирусных и плазмидных векторов. Мышей и собак с МПС I успешно лечили генной терапией. Большинство переносчиков могут корректировать заболевания печени и селезенки, а также могут корректировать последствия для мозга при высоких дозах. Генная терапия улучшила выживаемость, неврологические и физические симптомы; однако у некоторых животных развивались необъяснимые опухоли печени. Если вопросы безопасности будут решены, генная терапия может стать альтернативным методом лечения заболеваний МПС у людей в будущем. [11]
Sangamo Therapeutics со штаб-квартирой в Ричмонде, штат Калифорния , в настоящее время проводит клинические испытания, включающие редактирование генов с использованием нуклеазы цинковых пальцев (ZFN) для лечения МПС I. [12]
История
[ редактировать ]В 1919 году немецкий педиатр Гертруда Хёрлер описала синдром, включающий помутнение роговицы, аномалии скелета и умственную отсталость. Похожая болезнь «гаргулизм» была описана в 1917 году Чарльзом А. Хантером. Херлер не упомянул статью Хантера. Из-за перебоев в связи, вызванных Первой мировой войной , вполне вероятно, что она не знала о его исследовании. Синдром Гурлера теперь относится к МПС IH, а синдром Хантера — к МПС II. [13] [14] В 1962 году Шейе выявил более легкую форму МПС I, что привело к определению синдрома Шейе. [4]
См. также
[ редактировать ]- Синдром Хантера (МПС II)
- Синдром Санфилиппо (МПС III)
- Синдром Моркио (МПС IV)
- Синдром Марото-Лами (МПС VI)
Ссылки
[ редактировать ]- ^ Джеймс В.Д., Бергер Т.Г. и др. (2006). Болезни кожи Эндрюса: клиническая дерматология . Сондерс Эльзевир. ISBN 978-0-7216-2921-6 .
- ^ Jump up to: а б «Информационный бюллетень по мукополисахаридозам» . Национальный институт неврологических расстройств и инсульта . 15 ноября 2017 г. Проверено 11 мая 2018 г.
- ^ Майер CM (июль 1991 г.). «Обструкция дыхательных путей при синдроме Гурлера - рентгенографические особенности». Международный журнал детской оториноларингологии . 22 (1): 91–6. дои : 10.1016/0165-5876(91)90101-г . ПМИД 1917344 .
- ^ Jump up to: а б с Мур Д., Коннок М.Дж., Рэйт Э., Лавери С. (сентябрь 2008 г.). «Распространенность и выживаемость при мукополисахаридозе I: синдромы Гурлера, Гурлера-Шейе и Шейе в Великобритании» . Сиротский журнал редких заболеваний . 3:24 . дои : 10.1186/1750-1172-3-24 . ПМЦ 2553763 . ПМИД 18796143 .
- ^ Jump up to: а б с Баниказеми М. (12 октября 2014 г.). «Синдром Гурлера, синдром Гурлера-Шейе и синдром Шейе (мукополисахаридоз типа I)» . Медскейп . Проверено 10 мая 2018 г.
- ^ Чкиуа Л., Будабус Х., Джабалли И., Грисса О., Туркиа Х.Б., Тебиб Н., Ларади С. (май 2018 г.). «Новая мутация гена IDUA в сайте сплайсинга у тунисских родословных с синдромом Гёрлера» . Диагностическая патология . 13 (1). BioMed Central : 35. doi : 10.1186/s13000-018-0710-3 . ПМЦ 5975427 . ПМИД 29843745 .
- ^ «Ген IDUA» . Домашний справочник по генетике. 11 июня 2019 года . Проверено 18 июня 2019 г.
- ^ «Мукополисахаридоз I типа» . Домашний справочник по генетике . Проверено 10 мая 2018 г.
- ^ Питерс С., Шапиро Э.Г., Андерсон Дж., Хенсли-Дауни П.Дж., Клемперер М.Р., Коуэн М.Дж. и др. (апрель 1998 г.). «Синдром Гурлера: II. Результаты трансплантации костного мозга HLA-генотипически идентичного брата и HLA-гаплоидентичного донора у пятидесяти четырех детей. Совместная исследовательская группа по болезням хранения» . Кровь . 91 (7): 2601–8. дои : 10.1182/blood.V91.7.2601 . ПМИД 9516162 .
- ^ Стаба С.Л., Эсколар М.Л., По М., Ким Ю., Мартин П.Л., Сабольч П. и др. (май 2004 г.). «Трансплантация пуповинной крови от неродственных доноров пациентам с синдромом Гурлера» . Медицинский журнал Новой Англии . 350 (19): 1960–9. doi : 10.1056/NEJMoa032613 . PMID 15128896 . S2CID 43572313 .
- ^ Подумайте КП, Хаскинс М.Э. (сентябрь 2007 г.). «Генная терапия мукополисахаридоза» . Экспертное мнение о биологической терапии . 7 (9): 1333–45. дои : 10.1517/14712598.7.9.1333 . ПМК 3340574 . ПМИД 17727324 .
- ^ «Исследование возрастающей дозы редактирования генома с помощью терапевтической нуклеазы цинковых пальцев (ZFN) SB-318 у субъектов с МПС I» . www.clinicaltrials.gov . Национальная медицинская библиотека США . Проверено 7 февраля 2019 г.
- ^ Синдром Херлера в журнале Who Named It?
- ^ Херлер, Г. (1919). «О виде множественных отклонений преимущественно костной системы» . Журнал педиатрии . 24 (5–6): 220–234. дои : 10.1007/BF02222956 . S2CID 34471544 .