Синдром Хантера
| Синдром Хантера | |
|---|---|
 | |
| Пациент с синдромом Хантера | |
| Специальность | Эндокринология |
| Симптомы | Скелетные аномалии, потеря слуха, дегенерация сетчатки, увеличение печени и селезенки. |
| Осложнения | Заболевания верхних дыхательных путей; сердечно-сосудистая недостаточность |
| Причины | Дефицит фермента идуронат-2-сульфатазы. |
| Дифференциальный диагноз | Мукополисахаридоз I типа ; другие мукополисахаридозы |
| Прогноз | В тяжелых случаях смерть обычно наступает к 15 годам. В тяжелых случаях пациенты могут дожить до 50 лет. |
| Частота | 1 из 100 000–150 000 рождений мужского пола [1] |
Синдром Хантера , или мукополисахаридоз типа II ( МПС II ), представляет собой редкое генетическое заболевание , при котором большие молекулы сахара, называемые гликозаминогликанами (или ГАГ, или мукополисахариды), накапливаются в тканях организма. Это форма лизосомальной болезни накопления . Синдром Хантера вызван дефицитом лизосомального фермента идуронат-2-сульфатазы (I2S). [2] [3] Недостаток этого фермента приводит гепарансульфата и дерматансульфата во всех тканях организма. к накоплению [4] Синдром Хантера — единственный синдром МПС , демонстрирующий Х-сцепленное рецессивное наследование. [4]
Симптомы синдрома Хантера сравнимы с симптомами МПС I. Он вызывает нарушения во многих органах, включая скелет, сердце и дыхательную систему. В тяжелых случаях это приводит к смерти в подростковом возрасте. В отличие от MPS I, помутнение роговицы не связано с этим заболеванием. [1]
Признаки и симптомы
[ редактировать ]Синдром Хантера может проявляться множеством фенотипов . Традиционно его классифицируют как «легкое» или «тяжелое» в зависимости от наличия симптомов со стороны центральной нервной системы , но это чрезмерное упрощение. Пациенты с «ослабленными» или «легкими» формами заболевания все еще могут иметь серьезные проблемы со здоровьем. Для тяжелобольных пациентов клиническое течение относительно предсказуемо; пациенты обычно умирают в раннем возрасте. Для людей с более легкими формами заболевания существует более широкий спектр исходов. Многие доживают до 20-30 лет, но у некоторых продолжительность жизни может быть почти нормальной. Нарушения со стороны сердца и дыхания являются обычной причиной смерти пациентов с более легкими формами заболевания. [2]
Симптомы синдрома Хантера (МПС II) обычно не проявляются при рождении. Часто первые симптомы могут включать грыжи живота , ушные инфекции , насморк и простуду . Поскольку накопление ГАГ продолжается во всех клетках организма, признаки МПС II становятся более заметными. Внешний вид многих детей с синдромом включает характерную грубость черт лица, в том числе выдающийся лоб , нос со сплюснутой переносицей и увеличенный язык . У них также может быть большая голова и увеличенный живот. В тяжелых случаях МПС II диагноз часто ставится в возрасте от 18 до 36 месяцев. В более легких случаях пациенты проявляют себя так же, как дети с синдромом Гурлера-Шейе , и диагноз обычно ставится в возрасте от 4 до 8 лет. [2]
Продолжительное хранение ГАГ приводит к нарушениям во многих системах органов. Через 18 месяцев у детей с тяжелой формой МПС II может наблюдаться снижение развития и прогрессирующая потеря навыков. [1] Утолщение сердечных клапанов и стенок сердца может привести к прогрессирующему снижению сердечной функции. Стенки дыхательных путей также могут утолщаться, что приводит к обструктивному заболеванию дыхательных путей . Поскольку печень и селезенка со временем увеличиваются в размерах, живот может раздуться , что сделает грыжи более заметными. все основные суставы МПС II может поражать , что приводит к скованности суставов и ограничению движений. пальцев и большого пальца Прогрессирующее поражение суставов приводит к снижению способности поднимать мелкие предметы. Воздействие на другие суставы, такие как бедра и колени, может затруднить нормальную ходьбу. Если развивается синдром запястного канала , может произойти дальнейшее снижение функции руки. Могут поражаться сами кости, что приводит к низкому росту. Кроме того, каменистые поражения у некоторых людей на плечах, ногах и верхней части спины могут наблюдаться кожи цвета слоновой кости. Эти поражения кожи считаются патогномоничными для заболевания. Наконец, хранение ГАГ в головного мозга может привести к задержке развития с последующей умственной отсталостью и прогрессирующей потерей функций. [ нужна ссылка ]
Возраст появления симптомов и наличие или отсутствие поведенческих нарушений являются прогностическими факторами конечной тяжести заболевания у очень молодых пациентов. Поведенческие нарушения часто могут имитировать комбинации симптомов синдрома дефицита внимания и гиперактивности , аутизма , обсессивно-компульсивного расстройства и/или расстройства сенсорной обработки , хотя наличие и уровень симптомов различаются у каждого пострадавшего ребенка. К ним часто также относится отсутствие соответствующего чувства опасности и агрессия. Поведенческие симптомы МПС II обычно предшествуют нейродегенерации и часто усиливаются до тех пор, пока умственные нарушения не станут более выраженными. [5] К моменту смерти большинство детей с тяжелой формой МПС II имеют тяжелые умственные нарушения и полностью зависят от опекунов. [2]
Генетика
[ редактировать ]
Поскольку синдром Хантера является Х-сцепленным рецессивным заболеванием, ему преимущественно подвержены пациенты мужского пола. Ген IDS расположен на Х-хромосоме. Ген IDS кодирует фермент под названием идуронат-2-сульфатаза (I2S). Недостаток этого фермента приводит к накоплению ГАГ, вызывающих симптомы МПС II. [6]
Если женщина унаследует одну копию мутантного аллеля MPS II, у нее обычно будет нормальная копия гена IDS , которая может компенсировать мутантный аллель. Это известно как генетический носитель . Однако мужчина, унаследовавший дефектную Х-хромосому, обычно не имеет другой Х-хромосомы, которая могла бы компенсировать мутантный ген. Таким образом, для развития МПС II женщине необходимо унаследовать два мутантных гена, тогда как пациенту мужского пола достаточно унаследовать только один мутантный ген. Женщина-носитель может пострадать из-за Х-инактивации , которая является случайным процессом. [ нужна ссылка ]
Патофизиология
[ редактировать ]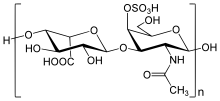
Человеческое тело зависит от огромного количества биохимических реакций, необходимых для поддержания важнейших функций. Одной из таких функций является распад крупных биомолекул . Нарушение этого процесса является основной проблемой синдрома Хантера и связанных с ним нарушений памяти. [ нужна ссылка ]
Биохимия синдрома Хантера связана с проблемами в части соединительной ткани, известной как внеклеточный матрикс , который состоит из различных сахаров и белков . Это помогает сформировать архитектурный каркас тела. Матрица окружает клетки тела организованной сетью и действует как клей, скрепляющий клетки тела вместе. Одной из частей внеклеточного матрикса является молекула, называемая протеогликан . Как и многие компоненты организма, протеогликаны необходимо расщеплять и заменять. Когда организм расщепляет протеогликаны, одним из образующихся продуктов являются мукополисахариды (ГАГ). [ нужна ссылка ]
При МПС II проблема касается распада двух ГАГ: дерматансульфата и гепарансульфата . На первом этапе расщепления дерматансульфата и гепарансульфата необходим лизосомальный фермент идуронат-2-сульфатаза, или I2S. У людей с МПС II этот фермент частично или полностью неактивен. В результате ГАГ накапливаются в клетках по всему организму, особенно в тканях, которые содержат большое количество дерматансульфата и гепарансульфата. Скорость накопления ГАГ не одинакова для всех людей с МПС II, что приводит к широкому спектру медицинских проблем. [ нужна ссылка ]
Диагностика
[ редактировать ]Первым лабораторным скрининговым тестом на расстройство МПС является анализ мочи на ГАГ. Аномальные значения указывают на вероятность расстройства МПС. Анализ мочи иногда может быть нормальным, даже если у ребенка действительно имеется расстройство МПС. Окончательный диагноз МПС II устанавливается путем измерения активности I2S в сыворотке , лейкоцитах или фибробластах кожи при биопсии . У некоторых людей с МПС II анализ гена может I2S определить тяжесть клинической картины. [ нужна ссылка ]
Пренатальная диагностика обычно доступна путем измерения ферментативной активности I2S в амниотической жидкости или в ткани ворсинок хориона . Если известно, что в семье встречается конкретная мутация, пренатальное молекулярно- генетическое тестирование можно провести . Секвенирование ДНК может выявить, является ли кто-то носителем заболевания. [2]
Уход
[ редактировать ]Из-за большого разнообразия фенотипов лечение этого расстройства подбирается индивидуально для каждого пациента. До недавнего времени эффективной терапии МПС II не существовало, поэтому паллиативная помощь применялась . Однако недавние достижения привели к созданию лекарств, которые могут улучшить выживаемость и благополучие людей с МПС II. [ нужна ссылка ]
Ферментозаместительная терапия
[ редактировать ]Идурсульфаза , очищенная форма недостающего лизосомального фермента, прошла клинические испытания в 2006 году. [6] США и впоследствии был одобрен Управлением по контролю за продуктами и лекарствами в качестве ферментозаместительного лечения МПС II. Идурсульфаза бета, еще один ферментозаместительный препарат, был одобрен в Корее Министерством безопасности пищевых продуктов и лекарств .
последние достижения в области заместительной ферментной терапии Доказано, что (ФЗТ) идурсульфазой улучшают многие признаки и симптомы МПС II, особенно если начать ее на ранних стадиях заболевания. После введения его можно транспортировать в клетки для расщепления ГАГ, но, поскольку лекарство не может проникнуть через гематоэнцефалический барьер , ожидается, что оно не приведет к улучшению когнитивных функций у пациентов с тяжелыми симптомами со стороны центральной нервной системы. Даже при ФЗТ необходимо лечение различных проблем органов у самых разных медицинских специалистов. [2]
Трансплантация костного мозга и стволовых клеток
[ редактировать ]трансплантация костного мозга и трансплантация гемопоэтических стволовых клеток (ТГСК). В некоторых исследованиях в качестве лечения использовались [7] [8] Хотя трансплантация принесла пользу многим системам органов, не было доказано, что она улучшает неврологические симптомы заболевания. Хотя ТГСК показала многообещающие результаты в лечении других расстройств МПС, ее результаты при лечении МПС II до сих пор были неудовлетворительными. Было показано, что ФЗТ приводит к лучшим результатам у пациентов с МПС II. [2]
Терапия редактирования генов
[ редактировать ]В феврале 2019 года ученые-медики, работающие с Sangamo Therapeutics со штаб-квартирой в Ричмонде , штат Калифорния, объявили о первой терапии «внутреннего» редактирования генов человека , позволяющей навсегда изменить ДНК – у пациента с МПС II. [9] Клинические испытания Sangamo, связанные с редактированием генов с использованием нуклеазы цинковых пальцев, продолжаются по состоянию на февраль 2019 года. [10]
Прогноз
[ редактировать ]Более раннее появление симптомов связано с худшим прогнозом. У детей, у которых проявляются симптомы в возрасте от 2 до 4 лет, смерть обычно наступает в возрасте от 15 до 20 лет. Причиной смерти обычно являются неврологические осложнения, обструктивное заболевание дыхательных путей и сердечная недостаточность. Если у пациентов минимальное неврологическое поражение, они могут дожить до 50 лет и старше. [1] [6]
Эпидемиология
[ редактировать ]По оценкам, во всем мире МПС II имеют 2000 человек, 500 из которых живут в Соединенных Штатах. [11]
Исследование, проведенное в Соединенном Королевстве, показало, что заболеваемость среди мужчин составляет примерно один из 130 000 живорожденных мальчиков. [12]
История
[ редактировать ]Синдром назван в честь врача Чарльза А. Хантера (1873–1955), который впервые описал его в 1917 году. [13] [14]
Исследовать
[ редактировать ]Начиная с 2010 года, в ходе клинических испытаний фазы I/II оценивались интратекальные инъекции более концентрированной дозы идурсульфазы, чем внутривенная форма, используемая при инфузиях заместительной ферментной терапии, в надежде предотвратить снижение когнитивных функций, связанное с тяжелой формой заболевания. [15] Результаты были объявлены в октябре 2013 года. [16] Клинические испытания фазы II/III начались в 2014 году. [17]
В 2017 году 44-летний мужчина [18] Пациент с МПС II лечился с помощью генной терапии в попытке предотвратить дальнейшее повреждение этой болезнью. Это первый случай применения редактирования генов у людей in vivo . [19] В 2018 году исследование было расширено до шести пациентов. [20]
Общество
[ редактировать ]24 июля 2004 года 38-летний Эндрю Рэгг из Уортинга , Западный Суссекс, Англия, задушил своего 10-летнего сына Джейкоба подушкой из-за инвалидности мальчика, связанной с MPS II. по военной безопасности Специалист Рэгг также заявил, что после возвращения с войны в Ираке он находился в состоянии стресса . Он отрицал убийство Джейкоба, но признал себя виновным в непредумышленном убийстве по причине ограниченной дееспособности. Судья Энн Рафферти назвала это дело «исключительным», приговорила Рэгга к двум годам тюремного заключения за непредумышленное убийство, а затем условила его приговор на два года. Рафферти сказал, что «ничего [не] можно было бы получить» от отправки Рэгга в тюрьму за это преступление. [21] [22] [23]
См. также
[ редактировать ]- Синдром Гурлера ( МПС I )
- Синдром Санфилиппо (МПС III)
- Синдром Моркио (МПС IV)
- Пренатальное тестирование
- Генетическое консультирование
Ссылки
[ редактировать ]- ^ Перейти обратно: а б с д «Информационный бюллетень по мукополисахаридозам» . Национальный институт неврологических расстройств и инсульта . 15 ноября 2017 года . Проверено 11 мая 2018 г.
- ^ Перейти обратно: а б с д и ж г Рэйт Дж.Э., Скарпа М., Бек М. и др. (март 2008 г.). «Мукополисахаридоз II типа (синдром Хантера): клинический обзор и рекомендации по лечению в эпоху заместительной ферментной терапии» . Евро. Ж. Педиатр . 167 (3): 267–77. дои : 10.1007/s00431-007-0635-4 . ПМК 2234442 . ПМИД 18038146 .
- ^ Джеймс, Уильям Д.; Бергер, Тимоти Г.; и др. (2006). Болезни кожи Эндрюса: клиническая дерматология . Сондерс Эльзевир. п. 544. ИСБН 978-0-7216-2921-6 .
- ^ Перейти обратно: а б Ле, Тао; Бхушан, Викас; Хофманн, Джеффри (2012). Первая помощь для USMLE Шаг 1 . МакГроу-Хилл . п. 117.
- ^ Шварц, Ида В.Д. (2007). «Клиническое исследование 77 больных мукополисахаридозом II типа». Акта Педиатрика . 96 (455): 63–70. дои : 10.1111/j.1651-2227.2007.00212.x . ПМИД 17391446 . S2CID 23119106 .
- ^ Перейти обратно: а б с Мюнцер, Дж; Рэйт, JE; Бек, М; Джулиани, Р; Харматц, П; Энг, КМ; Веллоди, А; Мартин, Р; Рамасвами, У; Гучавас-Чаликоглу, М; Виджаярагаван, С; Вендт, С; Пуга, AC; Ульбрих, Б; Шинави, М; Клири, М; Пайпер, Д; Конвей, AM; Кимура, А. (август 2006 г.). «Клиническое исследование фазы II/III ферментозаместительной терапии идурсульфазой при мукополисахаридозе II (синдром Хантера)» . Генетика в медицине . 8 (8): 465–73. дои : 10.1097/01.gim.0000232477.37660.fb . ПМИД 16912578 .
- ^ Гуффон, Н. (май 2009 г.). «Трансплантация костного мозга детям с синдромом Хантера: результат через 7–17 лет» . Журнал педиатрии . 154 (5): 733–737. дои : 10.1016/j.jpeds.2008.11.041 . ПМИД 19167723 .
- ^ Аннибали, Р. (октябрь 2013 г.). «Синдром Хантера (мукополисахаридоз типа II), тяжелый фенотип: долгосрочное наблюдение за пациентами, перенесшими трансплантацию гемопоэтических стволовых клеток». Том. 65, нет. 5. Минерва Педиатрика . стр. 487–496. ПМИД 24056375 .
- ^ Маркионе, Мэрилин (7 февраля 2019 г.). «Испытания показывают, что ученые впервые осуществили редактирование генов «внутри тела»» . АП Новости . Проверено 7 февраля 2019 г.
- ^ Персонал (2 февраля 2019 г.). «Исследование возрастающей дозы редактирования генома с помощью терапевтической нуклеазы цинковых пальцев (ZFN) SB-913 у субъектов с МПС II» . ClinicalTrials.gov . Национальная медицинская библиотека США . Проверено 7 февраля 2019 г.
- ^ LaTercera.com (на испанском языке) [ постоянная мертвая ссылка ]
- ^ Молодой ID, Харпер П.С. (1982). «Частота синдрома Хантера». Хм. Жене . 60 (4): 391–2. дои : 10.1007/BF00569230 . ПМИД 6809596 . S2CID 9667145 .
- ^ Синдром Хантера (Чарльз А. Хантер) в Who Named It?
- ^ Хантер, Калифорния (1917). «Редкая болезнь у двух братьев» . Труды Королевского медицинского общества . 10 (Секта Исследование Дитя). Лондон: 104–116. дои : 10.1177/003591571701001833 . ПМК 2018097 . ПМИД 19979883 .
- ^ «Рандомизированное исследование фазы I/II по безопасности и возрастанию дозы интратекального введения идурсульфазы-ИТ в сочетании с внутривенным введением элапразы у педиатрических пациентов с синдромом Хантера и когнитивными нарушениями» . Clinicaltrials.gov . Национальные институты здравоохранения США. 15 июня 2009 года . Проверено 22 июля 2018 г.
- ^ «Исследование безопасности и диапазона доз идурсульфазы (интратекального) введения через устройство для интратекальной доставки лекарств у педиатрических пациентов с синдромом Хантера, которые имеют поражение центральной нервной системы и получают лечение элапразой® - результаты» . Clinicaltrials.gov . Национальные институты здравоохранения США. 31 октября 2013 года . Проверено 20 июля 2014 г.
- ^ «Исследование интратекального введения идурсульфазы-IT в сочетании с Elaprase® у педиатрических пациентов с синдромом Хантера и ранними когнитивными нарушениями (AIM-IT)» . Clinicaltrials.gov . Национальные институты здравоохранения США. Июль 2014 года . Проверено 20 июля 2014 г.
- ^ Маркионе, Мэрилинн (15 ноября 2017 г.). «Американские ученые пробуют первое редактирование генов в организме» . Ассошиэйтед Пресс . Проверено 16 ноября 2017 г.
- ^ Маркионе, Мэрилин (14 ноября 2017 г.). «Ученые впервые попытались редактировать гены внутри пациента» . Время . Архивировано из оригинала 15 ноября 2017 года . Проверено 15 ноября 2017 г.
- ^ Маркионе, Майлинн (5 сентября 2018 г.). «Первые результаты вселяют надежды на историческую попытку редактирования генов» . АП Новости . Проверено 6 сентября 2018 г.
- ^ NEWS.BBC.co.uk , «Отец признан виновным в убийстве сына», BBC News
- ^ Guardian.co.uk , «Бывший солдат SAS, задушивший неизлечимо больного сына, выходит на свободу» The Guardian
- ^ NEWS.BBC.co.uk , «Обзор «прояснит законы об убийствах»» BBC News
Внешние ссылки
[ редактировать ] СМИ, связанные с синдромом Хантера, на Викискладе?
СМИ, связанные с синдромом Хантера, на Викискладе? - Запись GeneReview/NIH/UW о мукополисахаридозе типа II